


Forskerhold udvikler en universel og nøjagtig metode til at beregne, hvordan proteiner interagerer med lægemidler
Proteiner er store molekyler, der spiller en afgørende rolle i mange biologiske processer. De kan fungere som enzymer, der katalyserer kemiske reaktioner; receptorer, som binder til specifikke molekyler og udløser et cellulært respons; og transportører, som flytter molekyler over cellemembraner. Lægemidler virker ofte ved at binde sig til proteiner og forstyrre deres funktion.
Det kan dog være svært at forudsige, hvordan et lægemiddel vil interagere med et protein. Dette skyldes, at proteiner er komplekse molekyler med mange forskellige bindingssteder. Styrken af et lægemiddels binding til et protein afhænger af lægemidlets kemiske struktur, proteinets struktur og det miljø, hvori interaktionen finder sted.
AFE-metoden udviklet af UCSD-forskerne løser denne udfordring ved at bruge en kombination af beregnings- og eksperimentelle teknikker. Den beregningsmæssige komponent af metoden bruger en molekylær dynamik simulering til at beregne den frie energi af binding mellem et lægemiddel og et protein. Den eksperimentelle komponent af metoden bruger en teknik kaldet "fluorescensanisotropi" til at måle bindingsaffiniteten mellem et lægemiddel og et protein.
AFE-metoden er i stand til nøjagtigt at beregne bindingsaffiniteten af et lægemiddel til et protein, selv når proteinet er fleksibelt og har flere bindingssteder. Dette gør metoden til et værdifuldt værktøj til lægemiddelopdagelse.
"Vores metode kan hjælpe videnskabsmænd med at designe nye lægemidler, der er mere effektive og har færre bivirkninger," sagde Rommie Amaro, professor i kemi og biokemi ved UCSD og seniorforfatter af undersøgelsen. "Vi er spændte på at se, hvordan vores metode vil blive brugt til at udvikle nye behandlingsformer for sygdomme som kræft, Alzheimers og HIV."
Undersøgelsen blev offentliggjort i tidsskriftet Nature Methods.
 Varme artikler
Varme artikler
-
 Undersøgelse rapporterer første bevis på sociale relationer mellem chimpanser og gorillaerGrafisk abstrakt. Kredit:iScience (2022). DOI:10.1016/j.isci.2022.105059 En langtidsundersøgelse ledet af primatologen Crickette Sanz ved Washington University i St. Louis afslører det første bevis
Undersøgelse rapporterer første bevis på sociale relationer mellem chimpanser og gorillaerGrafisk abstrakt. Kredit:iScience (2022). DOI:10.1016/j.isci.2022.105059 En langtidsundersøgelse ledet af primatologen Crickette Sanz ved Washington University i St. Louis afslører det første bevis -
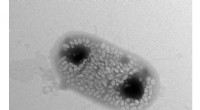 Forskere designer bakterier til at reflektere sonarsignaler til ultralydsbilleddannelseTransmission elektronmikrografi (TEM) billede af en enkelt kommensal bakterie, E coli Nissle 1917, som er blevet gensplejset til at udtrykke gasfyldte proteinnanostrukturer kendt som gasvesikler. Ce
Forskere designer bakterier til at reflektere sonarsignaler til ultralydsbilleddannelseTransmission elektronmikrografi (TEM) billede af en enkelt kommensal bakterie, E coli Nissle 1917, som er blevet gensplejset til at udtrykke gasfyldte proteinnanostrukturer kendt som gasvesikler. Ce -
 Fugle lærer af hinandens afsky, gør insekter i stand til at udvikle lyse farverStore tit ser videoen af en anden fugl, der oplever afsky som en del af undersøgelsen. Kredit:Liisa Hämäläinen Mange dyr har udviklet sig til at skille sig ud. Lyse farver er lette at få øje på,
Fugle lærer af hinandens afsky, gør insekter i stand til at udvikle lyse farverStore tit ser videoen af en anden fugl, der oplever afsky som en del af undersøgelsen. Kredit:Liisa Hämäläinen Mange dyr har udviklet sig til at skille sig ud. Lyse farver er lette at få øje på, -
 Hvordan hurtigtvoksende alger kan øge væksten af fødevareafgrøderNy forskning identificerer måder at øge afgrødeudbyttet ved at inkorporere strategier fra en hurtigtvoksende algeart i planter som hvede og ris. Kredit:Pixabay En ny undersøgelse giver en ramme for
Hvordan hurtigtvoksende alger kan øge væksten af fødevareafgrøderNy forskning identificerer måder at øge afgrødeudbyttet ved at inkorporere strategier fra en hurtigtvoksende algeart i planter som hvede og ris. Kredit:Pixabay En ny undersøgelse giver en ramme for
- Ny nanokrystallinsk legering, der kombinerer mekanisk styrke med krybningsmodstand ved høj temperat…
- 3D-trykt nanomateriale viser forskellige transparenter og farver
- I borofen, grænser er ingen barriere - forskere laver og tester atomtykke borer unikke domæner
- Bringe lægemidler til hjernen med nanopartikler til behandling af neurodegenerative sygdomme
- Hvordan man laver en løvfældende skov Diorama
- Pentagon kombinerer sødroner, AI til politiet i Golfregionen