


Et distribueret computerprojekt tager fat på COVID-19

Vincent Volz. Kredit:Temple University
Forskere over hele verden arbejder med hidtil uset hastighed og skala for at forstå coronavirus, udvikle en vaccine og opdage nye lægemidler til behandling af COVID-19.
Nu, borgere, allerede gør deres del for at stoppe virussens spredning gennem social distancering og andre foranstaltninger, kan også hjælpe med at udvikle nye terapier ved at køre simuleringer på deres computere. At udføre specifikke beregninger ved at koordinere og fordele arbejdet på tværs af tusindvis af separate computere kaldes distribueret computing.
Sammen med kandidatstuderende i sit laboratorium, Lektor i kemi Vincent Voelz har arbejdet sammen med et internationalt team af forskere på beregningsmæssigt at screene potentielle hæmmere af coronaviruss vigtigste protease, et attraktivt mål for nye antivirale lægemidler. Og de bruger det distribuerede computernetværk Folding@home til at gøre det. Folding refererer til de processer, hvorved en proteinstruktur antager sin form, så den kan udføre sine biologiske funktioner.
"Vores gruppe bruger værktøjerne til molekylær simulering og statistisk mekanik til at undersøge strukturen og funktionen af biomolekyler, " siger Voelz, som har arbejdet med Folding@home siden 2007, mens han var postdoc ved Stanford University, hvor det distribuerede computernetværk startede. "Det er et hurtigt spring fra det arbejde til at bruge vores ekspertise inden for biomolekylær simulering til at hjælpe med at bekæmpe COVID-19."
Til coronavirus-forskningen, Voelz samarbejder med forskere ved Memorial Sloan-Kettering Cancer Center og Diamond Light Source. En røntgenkrystallografigruppe i Storbritannien, Diamond Light Source har udført banebrydende arbejde med at løse mere end tusind forskellige krystalstrukturer af coronavirus hovedproteasen og opdage flere lægemiddelfragmenter, der binder til steder på proteinet.
"Når virussen kommer ind i en celle, den bruger cellens maskineri til at samle flere kopier af sig selv og replikere, " forklarer Voelz. "Hvis du kan hæmme proteasen, du kan hæmme et nødvendigt trin i virussens livscyklus."
Den kombinerede computerkraft fra Folding@homes tusindvis af brugere bliver brugt til nærmest at screene et stort antal potentielle lægemiddelforbindelser. Disse simuleringer vil hjælpe med at prioritere, hvilke molekyler der vil blive syntetiseret og analyseret af forskere, der sigter mod hurtigt at udvikle nye terapier mod coronavirus.
"Vi ved nu, at der er mange lægemiddelfragmenter, der binder sig til bestemte steder på coronaviruss proteinstruktur, " siger Voelz. "Dette er spor til yderligere lægemiddeludvikling. Den dynamiske information, vi får fra Folding@home-simuleringerne, er virkelig svær at måle eksperimentelt i et laboratorium."
I begyndelsen af marts, omkring 30, 000 brugere havde downloadet Folding@home-softwaren og var aktive deltagere i COVID-19-projektet. Flere uger senere, det tal var vokset til mere end 700, 000. Pr. 1. april der deltog mere end 1 million mennesker. "Kombineret, vi er nu den største supercomputer i verden, " siger Voelz, der bemærker, at online gaming-fællesskabet er en stor bidragyder. "Vi har brudt exaFLOP-barrieren, en måling af operationer per sekund, der svarer til ti gange regnekraften fra verdens hurtigste supercomputer."
Ifølge Voelz, hastigheden af coronaviruss spredning rundt i verden har inspireret mange forskere til at fjerne "flaskehalse" i, hvordan videnskabelig viden udvikles, analyseret og delt.
"Videnskabelige organisationer deler information på en hidtil uset måde, og mennesker rundt om i verden slår sig sammen for at løse et meget vanskeligt problem, " siger Voelz. "Folding@homes slags borgervidenskab eller crowdsourced videnskab kan være meget kraftfuld. Jo flere mennesker bliver tændt på denne idé, jo vigtigere grundlæggende videnskab kan vi lave."
Vil du hjælpe med at finde nye lægemidler til at bekæmpe COVID-19? Gå til foldingathome.org for at downloade softwaren.
 Varme artikler
Varme artikler
-
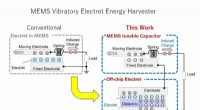 Udnytter gratis energi med små energihøstereI modsætning til konventionelle elektretbaserede MEMS energihøstere, som indeholder hele systemet i en enkelt chip, den foreslåede designmetodologi involverer at have elektreten og MEMS-afstembare kon
Udnytter gratis energi med små energihøstereI modsætning til konventionelle elektretbaserede MEMS energihøstere, som indeholder hele systemet i en enkelt chip, den foreslåede designmetodologi involverer at have elektreten og MEMS-afstembare kon -
 Boeing 737 MAX nødlander under amerikansk overførsel:FAAEt Southwest Airlines Boeing 737 MAX 8-fly – som disse afbilledet i Phoenix, Arizona tidligere på måneden - nødlandede den 26. marts, 2019, da den blev færget til Californien til opbevaring Et Boe
Boeing 737 MAX nødlander under amerikansk overførsel:FAAEt Southwest Airlines Boeing 737 MAX 8-fly – som disse afbilledet i Phoenix, Arizona tidligere på måneden - nødlandede den 26. marts, 2019, da den blev færget til Californien til opbevaring Et Boe -
 Ubers nye On-Trip Reporting-værktøj lader dig rapportere ubehagelige ture i realtidKredit:CC0 Public Domain Uber har lige tilføjet et nyt værktøj til ryttere, der befinder sig i ubehagelige situationer under en tur. Den tur-hyldende gigant lancerede On-Trip Reporting onsdag, so
Ubers nye On-Trip Reporting-værktøj lader dig rapportere ubehagelige ture i realtidKredit:CC0 Public Domain Uber har lige tilføjet et nyt værktøj til ryttere, der befinder sig i ubehagelige situationer under en tur. Den tur-hyldende gigant lancerede On-Trip Reporting onsdag, so -
 PayPal køber betalingsstart iZettle for $2,2 miaUSA-baseret PayPal, en enhed af eBay, har foretaget sit hidtil største opkøb ved at købe den svenske online-handelsstartup iZettle for 2,2 mia. PayPal annoncerede torsdag en aftale om at købe den
PayPal køber betalingsstart iZettle for $2,2 miaUSA-baseret PayPal, en enhed af eBay, har foretaget sit hidtil største opkøb ved at købe den svenske online-handelsstartup iZettle for 2,2 mia. PayPal annoncerede torsdag en aftale om at købe den
- Generel beskrivelse sætter gang i fremskridt inden for farvestofkemi
- Savner skoven for træerne:Et uventet billede af New York City -skove
- Brug af kunstig intelligens til at generere 3D-hologrammer i realtid
- Hvordan påvirker nitrogendynamikken kulstof- og vandbudgetter i Kina?
- Japans Takeda øjner overtagelsen af Shire
- Hvordan går undervands vulkaner ud?