


Det strukturelle mysterium med scandiumfluorid illustreret

Kredit:University of Connecticut
Den, der sagde, at reglerne skulle brydes, var ikke fysiker. Når noget ikke virker, som du tror, det burde, enten er reglerne forkerte, eller der er ny fysik, der skal opdages. Hvilket er præcis, hvad UConns Connor Occhialini, en senior æresstuderende med hovedfag i fysik og matematik, fundet, da han begyndte at forske i scandiumfluorid.
Scandium fluorid er en gennemsigtig krystal med en kubisk form, et biprodukt af minedrift. Det bruges ikke kommercielt, og det ville ikke være specielt interessant for nogen bortset fra en mærkelig ting:det krymper, når det varmer.
De fleste materialer svulmer, når de opvarmes. Virkelig simple materialer som brintgas svulmer, fordi varmen får deres atomer til at zoome hurtigere rundt, støder mere ind i hinanden, så det samme antal brintatomer har brug for mere plads. Mere komplicerede materialer svulmer også op, derfor har din trædøre en tendens til at sidde fast om sommeren. Men faste stoffer som træ kan ikke svulme så meget som en gas, fordi deres atomer er tæt forbundet til lange, sammenlåste molekyler, så de bare tumler rundt, hævede døren en lille smule.
Scandium fluorid må gøre noget andet, ræsonnerede Occhialini. Hans rådgiver for hans æresfysikprojekt, Jason Hancock, havde arbejdet med scandiumfluorid, og bad Occhialini om at studere en model af krystallens dynamik. Scandiumfluorid har en ret simpel struktur:det er en solid krystal, med hvert skandiumatom omgivet af seks fluorstoffer for at lave stakke af oktaedre (ottesidede diamanter). Forskerne håbede, at den simple struktur kunne være let at forstå. Forståelse af scandiumfluorids mærkelige 'negative termiske udvidelse, ' som fysikere kalder det varmerelaterede svind, kan give mere generel indsigt i andre, mere komplekse materialer, der gør det samme.

Figur 1. Hjælp, jeg krymper! De sorte diamanter repræsenterer scandiumfluoridmolekyler. Mens de varmer, de roterer, og krystallerne trækker sig sammen. Læg mærke til, hvordan molekylerne nær massecentrum (central prik) bevæger sig mindre end molekylerne tættere på krystallens kant. Kredit:University of Connecticut
Occhialinis første skridt var at forenkle problemet. Så i stedet for en tredimensionel krystal, han besluttede at tænke på det som et todimensionelt ark.
Hver sort diamant repræsenterer et molekyle af scandiumfluorid. Scandium-atomerne (blå prikker) er i midten af hver diamant, og et fluoratom er i hvert hjørne.
Det meste af tiden, bindinger mellem atomer er fleksible. Så i et normalt krystallinsk fast stof – calciumfluorid, for eksempel – ville fluor- og calciumatomerne alle være i stand til at vrikke rundt uafhængigt, når materialet varmes op. Mens de vrikkede, de ville fylde lidt mere, og det faste stof ville svulme. Normal solid adfærd.
Men Occhialini spekulerede på, om det måske ikke var det, der skete i scandiumfluorid. Måske i denne model, han skulle antage, at bindingerne, der forbinder hver fluor til dens skandium, var stive? Så stive bevæger fluor-skandium-bindingerne sig slet ikke, så diamanterne er som massive blokke. De eneste steder, hvor strukturen ville være i stand til at bøje sig, når den varmes op, ville være ved fluoratomerne, som ville fungere som små små led. Mens krystallen blev varmet op, de små scandiumfluoridblokke ville vippe rundt om fluorstofferne i hjørnerne. Det er det, du ser ske på billedet. Du vil bemærke, at når diamanterne vipper, hele strukturen bliver mindre. Det strammer faktisk op. Den blå kontur viser strukturen på det koldeste, perfekt ordnet tilstand, uden molekylær bevægelse. Når diamanterne vipper, de fylder et mindre samlet volumen, end det blå omrids afgrænser. Dette er negativ termisk udvidelse.
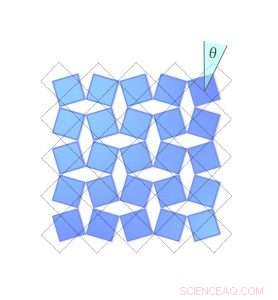
Figur 2. Hvor meget en scandiumfluorid krystal krymper afhænger af, hvor langt molekylerne roterer. Her, den blå diamant i øverste højre hjørne roterer med uret, fejer en vinkel theta ud. De stiplede linjer viser dens position, når vinklen var nul. Kredit:University of Connecticut
Occhialini fandt ud af, at du kan beskrive dette svind matematisk, kun ved at bruge vinklen på molekylernes hældning. Han kaldte vinklen Θ (theta). Når scandiumfluoridblokkene vipper med en vinkel Θ, afstanden mellem midten af hver blok forkortes med en faktor cosinus Θ, og krystallens samlede volumen krymper.
For at beregne dette svind (eller, i et normalt materiale, udvidelse) i detaljer, Occhialini føjede et tredje led til den klassiske ligning, der beskriver energien i en vibrerende krystal. De to første led i standardligningen beskriver den potentielle energi en krystal har fra bøjningen ved hver molekylær forbindelse, plus den kinetiske rotationsenergi for hvert molekyle. Occhialinis ligning beskriver også den translationelle kinetiske energi af molekylerne – ikke kun fra at rotere rundt, men bevæger sig også mod og væk fra deres oprindelige positioner, når de roterer. Jo længere de er fra krystallens massecenter, jo mere de bevæger sig. Se tilbage på figur 1 og læg mærke til prikken i midten; det er massens centrum. Diamanterne i midten bevæger sig næsten ikke i forhold til det, mens diamanterne i kanterne bevæger sig meget. Forestil dig nu, hvor stor en forskel der ville være, hvis krystallen havde millioner af molekyler i stedet for kun 25. Og nu forstår du, hvor vigtigt det tredje led kunne være for krystallens energi.
Nu, molekyler er molekyler, de skrumper ikke bare ind og bliver der. De bevæger sig konstant, og jo varmere de bliver, jo mere de bevæger sig. En del af Occhialinis indsigt er, at gennemsnitlig, den molekylære struktur bliver bøjere jo varmere det bliver. Så molekylerne vipper mere og bruger mere tid på større værdier af Θ, tættere på 45 grader. Efter at Occhialini havde tænkt over det et stykke tid sammen med Hancock og fysik-ph.d.-studerende Sahan Handunkanda og Erin Curry, de indså, at der var en geometrisk form, der havde den samme matematiske beskrivelse. Det er Archimedes' spiralpendul.
Hver drejning af spiralen er nøjagtig den samme afstand fra den sidste. Denne afstand – afstanden mellem svingene – styres af Θ. Forestil dig en linje, der strækker sig fra midten af kuglen til et punkt på spiralen. Vinklen mellem den linje og kuglens pol er Θ. Ser du den lille bold rejse langs spiralen? Det er enden på den imaginære linje. Efterhånden som Θ bliver større, bolden bevæger sig mod ækvator. Forestil dig, at kuglen repræsenterer den øjeblikkelige tilstand af scandiumfluorid-krystallen - fysikerne beregnede det statistiske gennemsnit af, hvad hvert molekyle i krystallen gør. Du vil bemærke, at bolden bruger mere tid i nærheden af ækvator i spiralkuglen, det er, den har en tendens til at hænge ud, hvor Θ er stor. Hvis temperaturen på krystallen falder, og molekylerne vrikker mindre, Θ bliver mindre, jo mere tid bolden tilbringer i nærheden af sfærens pol, og jo mindre krymper krystallen.
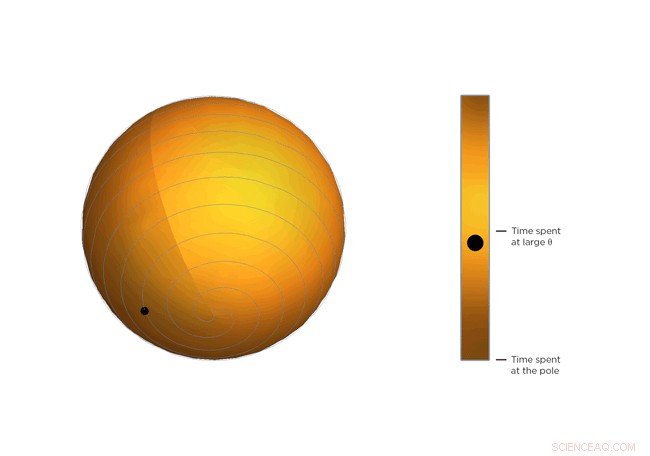
Figur 3. Vrid og krymp. Ligningen, der beskriver rotationen af scandiumfluorid-molekylerne, er den samme som ligningen, der beskriver bevægelsen af en kugle på en Archimedes' spiralpendul. Læg mærke til, hvordan den bruger mere tid i større vinkler. Kredit:University of Connecticut
Så ikke kun kan et virkelig mærkeligt fænomen med en krystal, der krymper, når den opvarmes, forklares ved blot at antage, at molekylerne er stive, men det kan illustreres med en klassisk geometrisk form!
Occhialini var blot en førsteårsstuderende, da Hancock introducerede ham til scandiumfluorid-puslespillet. Han måtte lære matematik, mens han gik, men efter omkring to semestres arbejde med det havde han fundet ud af ligningen, der beskrev, hvad der foregik. Nu på sit sidste år, han siger, at hans forskningserfaringer i Hancocks laboratorium har været en integreret del af hans erfaring som bachelor.
Ligningen fungerer smukt og forklarer også visse aspekter af Hancocks eksperimentelle røntgenmålinger.
"Jeg lærte meget mere at lave research, end noget kursus kunne have givet mig, " siger Occhialini.
 Varme artikler
Varme artikler
-
 Forskere opnår betydeligt gennembrud inden for topologiske isolator-baserede enheder til moderne sp…Figur:(a) Skematisk diagram, der illustrerer Dirac-keglen af topologisk isolator og spin-momentum-låsning. (b) Topologiske isolator/ferromagnet (Bi2Se3/NiFe) spin-orbit momentenheder. (c-e) Magneto-
Forskere opnår betydeligt gennembrud inden for topologiske isolator-baserede enheder til moderne sp…Figur:(a) Skematisk diagram, der illustrerer Dirac-keglen af topologisk isolator og spin-momentum-låsning. (b) Topologiske isolator/ferromagnet (Bi2Se3/NiFe) spin-orbit momentenheder. (c-e) Magneto- -
 Eksempler på enkle maskiner og komplekse maskinerEn maskine er et værktøj, der bruges til at gøre arbejdet lettere. Det kan gøre det ved at ændre en kraftretning, øge afstandens eller hastigheden for en styrke, overføre en kraft fra et sted til et a
Eksempler på enkle maskiner og komplekse maskinerEn maskine er et værktøj, der bruges til at gøre arbejdet lettere. Det kan gøre det ved at ændre en kraftretning, øge afstandens eller hastigheden for en styrke, overføre en kraft fra et sted til et a -
 Vender forvikling på hovedetKredit:CC0 Public Domain Et team af fysikere fra ICTP-Trieste og IQOQI-Innsbruck er kommet med en overraskende enkel idé til at undersøge kvanteindvikling af mange partikler. I stedet for at grave
Vender forvikling på hovedetKredit:CC0 Public Domain Et team af fysikere fra ICTP-Trieste og IQOQI-Innsbruck er kommet med en overraskende enkel idé til at undersøge kvanteindvikling af mange partikler. I stedet for at grave -
 Tumbling eller Cheerleading Science Fair ProjekterVidenskabelige elementer er meget lettere at forstå, når de er relateret til noget uden for tal, formler og teorier. Med alle bevægelser, danse, sang, stunts og tumbling er cheerleading en ekstrem fys
Tumbling eller Cheerleading Science Fair ProjekterVidenskabelige elementer er meget lettere at forstå, når de er relateret til noget uden for tal, formler og teorier. Med alle bevægelser, danse, sang, stunts og tumbling er cheerleading en ekstrem fys
- Nyt værktøj til at måle kønsbias på arbejdspladsen kan hjælpe med endelig at fjerne det
- Ossia klæder sig til en ny dag inden for trådløs opladning
- Brandmænd begrænser enorme naturbrande på Korsika og Portugal
- Papua Ny Guinea lukker forurenende kinesisk fabrik
- At brygge mesopotamisk øl bringer en tår af denne pulserende gamle drikkekultur til live igen
- NASA afslører finalister til næste New Frontiers robotmission:Saturns månen Titan eller Rosetta r…