


Ny teknik kan gøre modellering af molekyler meget lettere
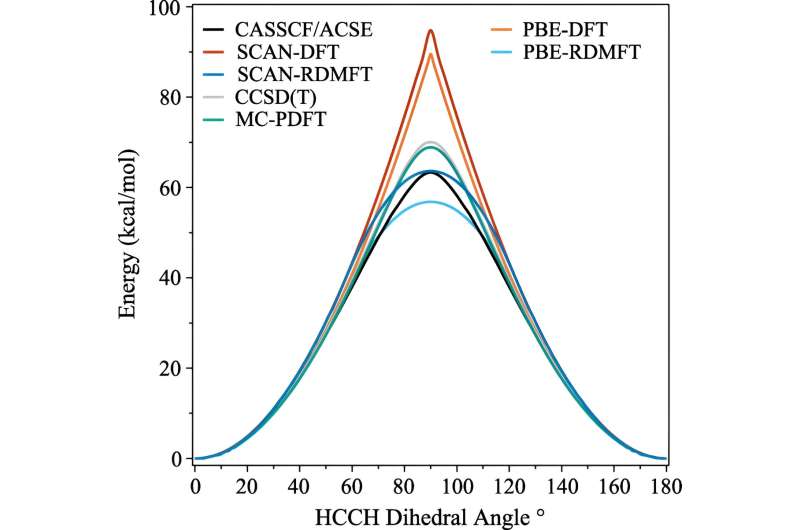
Meget ligesom de mennesker, der skabte dem, finder computere fysik hårdt, men kvantemekanik endnu sværere. Men en ny teknik skabt af tre forskere fra University of Chicago tillader computere at simulere visse udfordrende kvantemekaniske effekter i komplekse elektroniske materialer med langt mindre indsats.
Ved at gøre disse simuleringer mere nøjagtige og effektive håber forskerne, at teknikken kan hjælpe med at opdage nye molekyler og materialer, såsom nye typer solceller eller kvantecomputere.
"Dette fremskridt rummer et enormt potentiale for at fremme vores forståelse af molekylære fænomener, med betydelige implikationer for kemi, materialevidenskab og beslægtede områder," sagde videnskabsmand Daniel Gibney, en Ph.D. studerende i kemi og første forfatter på papiret, offentliggjort 14. december i Physical Review Letters .
Elektroner og energi
Et blad eller et solpanel ser glat og enkelt ud udefra, men zoom ned til det molekylære niveau, og du vil se en vildt kompliceret dans af elektroner og molekyler.
For at gøre nye fremskridt inden for bæredygtighed, fremstilling, landbrug og mange andre områder modellerer videnskabsmænd adfærden af disse kemiske og molekylære interaktioner. Dette hjælper med at afsløre nye designmuligheder for fremtiden – for alt fra nye måder at binde kuldioxid til nye typer kvantebits.
Mange fremskridt er blevet gjort i de sidste årtier, men et af de områder, der er forblevet stædigt vanskeligt at simulere, er, når molekylerne begynder at udvise kompleks kvantemekanisk adfærd, som forskerne kalder stærk korrelation.
Problemet er, at når først elektroner begynder at fremvise deres mest kvantemekaniske effekter - såsom at blive "viklet ind" - har beregningerne øjeblikkeligt brug for meget mere computerkraft. Selv supercomputere kæmper for at håndtere konsekvenserne.
En af de almindeligt anvendte beregninger kaldes tæthedsfunktionsteori. "Dette er dybest set den mest allestedsnærværende teknik til at forudsige elektronisk struktur, men det er i bund og grund en tilnærmelse, hvor alle elektronerne behandles som en funktion af en elektron," forklarede David Mazziotti, professor i kemi og seniorforfatter til undersøgelsen.
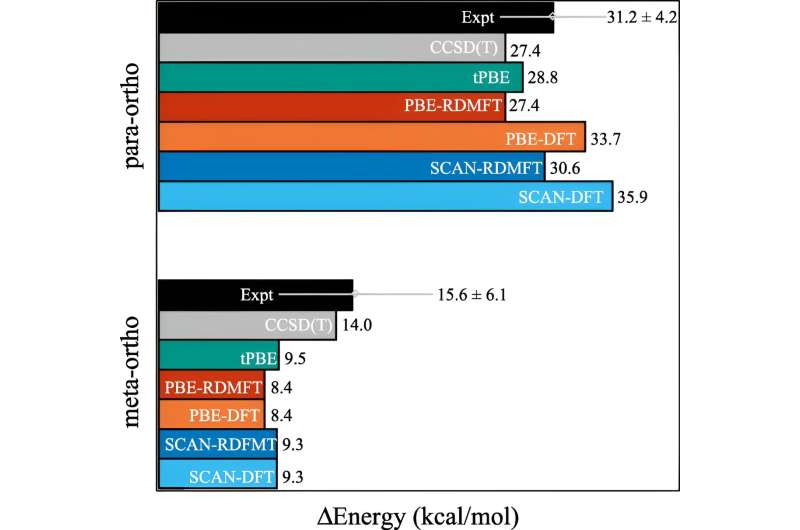
For mange beregninger gør en tilnærmelse jobbet. Men det begynder at bryde sammen, efterhånden som elektronernes adfærd bliver mere korreleret, som det sker, når kvantemekanikken begynder at spille ind. I kvantemekanikken kan disse elektroner være flere steder, eller orbitaler, samtidigt. Dette hæmmer ikke kun menneskelige hjerner, men også tæthedsfunktionsteori.
"Og dette er et vigtigt problem, fordi mange af de problemer, vi bekymrer os om i det 21. århundrede - som nye molekyler og materialer til vedvarende energi og bæredygtighed - kræver, at vi udnytter materialernes kvantenatur," sagde Mazziotti.
Mazziotti, Gibney og tredje forfatter Jan-Niklas Boyn fandt ud af, at de kunne tilføje en universel korrektion til densitetsfunktionsteori, der gør det muligt for elektronerne at blive viklet ind mellem flere orbitaler på én gang.
"Dette gør det muligt for orbitalerne i beregningen ikke kun at være helt fyldte eller helt tomme, men hvor som helst derimellem," sagde Mazziotti. "Vi når frem til et billede af én elektron, der stadig er i stand til at fange den adfærd, der opstår fra korrelerede elektroneffekter med mange krop."
En 'universel' tilpasning
Som en bonus, sagde forskerne, kan koden føjes til eksisterende algoritmer uden at skulle omskrive den kode. "Dybest set træder rettelsen i gang, når det er nødvendigt, men forstyrrer ellers ikke resten af koden," sagde Gibney.
Det er også universelt – idet det kan føjes til kode, der simulerer mange slags elektronisk adfærd, det være sig fotovoltaiske solpaneler eller kulstofbinding eller superledende materialer – eller endda biologi.
For eksempel, forklarede Boyn, kunne en anvendelse være at forstå den kemi, der finder sted ved hjælp af enzymer, der indeholder metalatomer, kendt som metalloenzymer.
"Der er et væld af metalloenzymer, der er ansvarlige for meget af kemien i dine celler, for eksempel, men de har været notorisk svære at beskrive med nuværende modeller," sagde han. "Denne teori kunne i den nærmeste fremtid give os mulighed for at tackle denne kemi på en måde, som er umulig lige nu."
Flere oplysninger: Daniel Gibney et al., Universal Generalization of Density Functional Theory for Static Correlation, Physical Review Letters (2023). DOI:10.1103/PhysRevLett.131.243003
Journaloplysninger: Physical Review Letters
Leveret af University of Chicago
 Varme artikler
Varme artikler
-
 Visualisering af elektriske mønstre, der ligger til grund for unormale hjertesammentrækninger og d…Driften og responsen mellem det virtuelle hjerte (system 1) og det modellerede arytmiske hjerte (system 2). Kredit:University Medical Center Göttingen På trods af fremskridt inden for medicinsk bi
Visualisering af elektriske mønstre, der ligger til grund for unormale hjertesammentrækninger og d…Driften og responsen mellem det virtuelle hjerte (system 1) og det modellerede arytmiske hjerte (system 2). Kredit:University Medical Center Göttingen På trods af fremskridt inden for medicinsk bi -
 Kvanteberegning med hullerDe to huller er begrænset til det germanium-rige lag kun et par nanometer tykt. På toppen, de elektriske porte er dannet af individuelle ledninger med påført spændinger. De positivt ladede huller mærk
Kvanteberegning med hullerDe to huller er begrænset til det germanium-rige lag kun et par nanometer tykt. På toppen, de elektriske porte er dannet af individuelle ledninger med påført spændinger. De positivt ladede huller mærk -
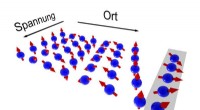 Manipulering af elektronspin uden tab af informationElektroner roterer på deres vej gennem chippen i et spiralmønster. Justeringer i spændingen fører til ændringer i bølgelængderne af dette mønster og dermed kan orienteringen af spindet kontrolleres.
Manipulering af elektronspin uden tab af informationElektroner roterer på deres vej gennem chippen i et spiralmønster. Justeringer i spændingen fører til ændringer i bølgelængderne af dette mønster og dermed kan orienteringen af spindet kontrolleres. -
 Forskere bygger transistorlignende port til kvanteinformationsbehandling - med quditsEn to-qudit-port, blandt de første af sin slags, maksimerer sammenfiltringen af fotoner, så kvanteinformation kan manipuleres mere forudsigeligt og pålideligt. Kredit:Purdue University billede/Allis
Forskere bygger transistorlignende port til kvanteinformationsbehandling - med quditsEn to-qudit-port, blandt de første af sin slags, maksimerer sammenfiltringen af fotoner, så kvanteinformation kan manipuleres mere forudsigeligt og pålideligt. Kredit:Purdue University billede/Allis
- Dækafgrøder giver bed and breakfast mellemlanding for trækfugle
- Varme, weekender, aggression og Chicago sommerskyderier
- Hvad er Factoring i Math?
- Budgetvenlige måder at få din veggie-fix på, når priserne stiger
- Nyt papir peger på jordporestruktur som nøglen til kulstoflagring
- Hvad er de tre tidsperioder, hvor dinosaurerne levede i?