


At se nærmere på genetiske skift i kræft
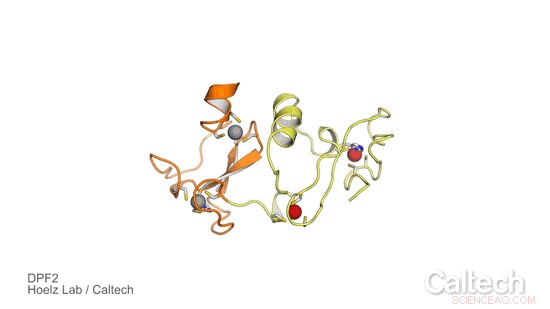
En krystalstruktur af en del af menneskelig DPF2, et protein, der styrer en genetisk switch, der fortæller blodstamceller, hvornår de skal blive røde og hvide blodlegemer. Orange og gule områder illustrerer DPF2 'læser'-domænet, som stabiliseres af zinkioner, repræsenteret som røde og grå kugler. Kredit:Hoelz Lab/Caltech
Mange ting går galt i cellerne under udviklingen af kræft. Kernen i kaosset er ofte genetiske afbrydere, der styrer produktionen af nye celler. I en særlig aggressiv form for leukæmi, kaldet akut myeloid leukæmi, en genetisk switch, der regulerer modningen af blodstamceller til røde og hvide blodlegemer, går galt. Normalt, dette skifte fører til passende antal hvide og røde blodlegemer. Men patienter med akut myeloid leukæmi ender med en farlig ophobning af blodstamceller og mangel på røde og hvide blodlegemer - celler, der er nødvendige for at forsyne kroppen med ilt og bekæmpe infektioner.
Nu, forskere ved Caltech og Sylvester Comprehensive Cancer Center ved University of Miami er ved at indsnævre et protein, der hjælper med at kontrollere denne genetiske switch. Hos raske personer, proteinet, kaldet DPF2, stopper produktionen af røde og hvide blodlegemer, når de ikke skal udskiftes. Det er, det slår kontakten fra. Men proteinet kan overproduceres hos patienter med akut myeloid leukæmi. Proteinet sidder dybest set på kontakten, forhindrer den i at tænde igen for at lave blodcellerne efter behov. Patienter, der overproducerer DPF2, har en særlig dårlig prognose.
I en ny undersøgelse, udkommer i ugen den 22. maj, 2017, i journalen Proceedings of the National Academy of Sciences , forskerne demonstrerer nye måder at hæmme DPF2, potentielt gør akut myeloid leukæmi mere behandlelig. De rapporterer nye strukturelle og funktionelle detaljer om et fragment af DPF2. Denne nye information afslører mål for udviklingen af lægemidler, der ville blokere proteinets funktion.
"Mange menneskelige sygdomme, herunder kræftformer, opstår på grund af funktionsfejl i genetiske kontakter, siger André Hoelz, den tilsvarende forfatter til undersøgelsen. Hoelz er professor i kemi ved Caltech, en Heritage Medical Research Institute (HMRI) efterforsker, og en Howard Hughes Medical Institute (HHMI) fakultetsstipendiat. "At belyse, hvordan de arbejder med atomare detaljer, giver os mulighed for at begynde processen med skræddersyede lægemidler for at inaktivere dem, og i mange tilfælde er det et vigtigt skridt mod en kur."
Røde og hvide blodlegemer regenereres konstant fra blodstamceller, som bor i vores knoglemarv. Ligesom andre stamceller, blodstamceller kan leve evigt. Det er først, når de bliver differentieret til specifikke celletyper, såsom røde og hvide blodlegemer, at de så bliver dødelige, eller erhverve evnen til at dø efter en vis periode.
"Vores kroppe bruger en kompleks række af genetiske skift til at differentiere en blodstamcelle til mange forskellige celletyper. Disse differentierede celler cirkulerer derefter i blodet og tjener en række forskellige funktioner. Når disse celler når slutningen af deres levetid, skal de udskiftes, " siger Hoelz. "Dette er lidt ligesom at udskifte brugte dæk på en bil."
For at undersøge DPF2's rolle og lære mere om, hvordan det styrer den genetiske switch til fremstilling af blodceller, Hoelz-gruppen samarbejdede med Stephen D. Nimer, co-korresponderende forfatter af papiret og direktør for Sylvester Comprehensive Cancer Center, og hans hold. Først, Ferdinand Huber og Andrew Davenport - begge kandidatstuderende ved Caltech i Hoelz-gruppen og co-first-forfattere af den nye undersøgelse - opnåede krystaller af en del af DPF2-proteinet, der indeholdt et domæne kendt som en PHD-finger, som står for planet homeodomain. De brugte derefter røntgenkrystallografi, en proces, der involverer at udsætte proteinkrystaller for højenergi røntgenstråler, at løse strukturen af PHD-fingerdomænet. Teknikken blev udført ved Stanford Synchrotron Radiation Lightsource, ved hjælp af en dedikeret beamline af Caltechs Molecular Observatory.
Resultaterne afslørede, hvordan DPF2 binder til et DNA-proteinkompleks, kaldet nukleosomet, at blokere produktionen af røde og hvide blodlegemer. Proteinet "læser" forskellige signaler vist på nukleosomoverfladen ved at antage en form, der passer til forskellige modifikationer på nukleosomkomplekset, ligesom de forskellige formede brikker i et puslespil. Når først proteinet binder til dette DNA-locus, DPF2 slår kontakten fra, der regulerer blodcelledifferentiering.
Det næste skridt var at se, om DPF2 kunne blokeres i menneskelige blodstamceller i laboratoriet. Sarah Greenblatt, en postdoc associeret i Nimers gruppe og medførsteforfatter af undersøgelsen, brugte den strukturelle information fra Hoelz' gruppe til at skabe en muteret version af proteinet. Nimer-gruppen introducerede derefter det muterede protein i blodstamceller, og fandt ud af, at den muterede DPF2 ikke længere kunne binde til nukleosomet. Med andre ord, DPF2 kunne ikke længere inaktivere kontakten til fremstilling af blodceller.
"Den muterede DPF2 var ude af stand til at binde sig til specifikke regioner i genomet og kunne ikke standse blodstamcelledifferentiering, " siger Huber. "Om DPF2 også kan blokeres i selve kræftpatienterne, skal vise sig." Forskerne siger, at en strukturel fatning i DPF2, et af de puslespilslignende områder identificeret i den nye undersøgelse, er et godt mål for kandidatlægemidler.
 Varme artikler
Varme artikler
-
 Elektronkrystallografi viste sig at virke lige så godt som røntgenkrystallografi kun på mindre kr…Identifikation af forbindelser fra heterogene blandinger. EM-gitter forberedt som ovenfor med biotin, brucine, carbamazepin, og cinchoninpulver blandet sammen. Alle fire forbindelser identificeret ved
Elektronkrystallografi viste sig at virke lige så godt som røntgenkrystallografi kun på mindre kr…Identifikation af forbindelser fra heterogene blandinger. EM-gitter forberedt som ovenfor med biotin, brucine, carbamazepin, og cinchoninpulver blandet sammen. Alle fire forbindelser identificeret ved -
 Hvordan nobelvindende kemikere brugte og styrede evolutionenDe amerikanske videnskabsmænd Frances Arnold og George Smith og den britiske forsker Gregory Winter har vundet Nobelprisen i kemi 2018 Tre videnskabsmænd delte onsdag Nobel Kemiprisen 2018 for der
Hvordan nobelvindende kemikere brugte og styrede evolutionenDe amerikanske videnskabsmænd Frances Arnold og George Smith og den britiske forsker Gregory Winter har vundet Nobelprisen i kemi 2018 Tre videnskabsmænd delte onsdag Nobel Kemiprisen 2018 for der -
 Verdens fineste guldprøve undersøgt med Los Alamos neutronerThe Rams Horn wire guldprøve. Kredit:Harvard University Ved at bruge neutronkarakteriseringsteknikker har et hold videnskabsmænd kigget ind i et af de mest unikke eksempler på trådguld, for første
Verdens fineste guldprøve undersøgt med Los Alamos neutronerThe Rams Horn wire guldprøve. Kredit:Harvard University Ved at bruge neutronkarakteriseringsteknikker har et hold videnskabsmænd kigget ind i et af de mest unikke eksempler på trådguld, for første -
 Nye syntetiske biologiværktøjer låser op for kompleks anlægsteknikEt blad med forskellige syntetiske promotorer, der udtrykker grønt fluorescerende protein, demonstrerer rækken af ekspressionsniveauer, der kan opnås med deres konstruerede promotorer. Kredit:JBEI/B
Nye syntetiske biologiværktøjer låser op for kompleks anlægsteknikEt blad med forskellige syntetiske promotorer, der udtrykker grønt fluorescerende protein, demonstrerer rækken af ekspressionsniveauer, der kan opnås med deres konstruerede promotorer. Kredit:JBEI/B
- Sådan beregnes en Rate Constant
- Hvordan fungerer en seismograf? Hvad er Richter -skalaen?
- Forskere udvikler nyt zoneinddelingsværktøj, der leverer globale topografiske datasæt på få min…
- Den første demonstration af fasetilpasning mellem en elektronbølge og en lysbølge
- NASA FDL udvikler nye tilgange til asteroide, komet- og soltrusler ved hjælp af kunstig intelligens
- Første plan nogensinde afsløret for at konstruere en kvantecomputer i stor skala