


Et frisk matematisk perspektiv åbner nye muligheder for beregningskemi
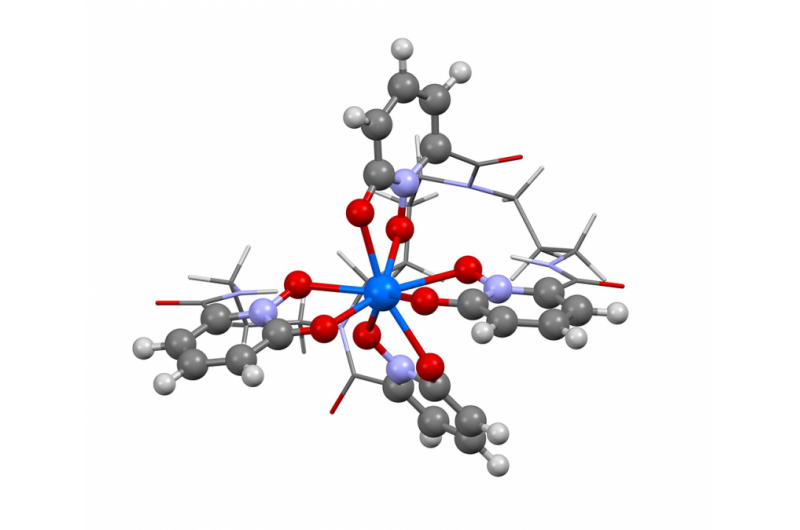
Dette billede viser strukturen af berkelium i oxidationstilstand +IV. Forskere brugte den nye Berkeley Lab-algoritme til at beregne absorptionsspektret og bekræfte, hvad flere eksperimentelle resultater har antydet - at grundstoffet berkelium bryder form med sine tunge grundstof-peers ved at påtage sig en ekstra positiv ladning, når det er bundet til et syntetisk organisk molekyle. Denne egenskab kan hjælpe forskere med at udvikle bedre metoder til håndtering og rensning af nukleare materialer. Kredit:Bert de Jong, Berkeley Lab
Objekter, der lyser i mørket, virker magiske, når du er barn - de kan lyse et mørkt rum op uden behov for elektricitet, batterier eller en pære. Så lærer du på et tidspunkt videnskaben bag dette fænomen. Kemiske forbindelser kaldet kromoforer får energi, eller spændt, når de absorberer synligt lys. Når de vender tilbage til deres normale tilstand, den lagrede energi frigives som lys, som vi opfatter som en glød. I materialevidenskab, forskere er afhængige af et lignende fænomen for at studere strukturerne af materialer, der i sidste ende vil blive brugt i kemisk katalyse, batterier, solenergi applikationer og mere.
Når et molekyle absorberer en foton - den grundlæggende partikel af lys - fremmes elektroner i det molekylære system fra en lavenergitilstand (jordtilstand) til en tilstand med højere energi (exciteret). Disse svar giver genlyd ved specifikke lysfrekvenser, efterlader "spektrale fingeraftryk", der belyser de atomare og elektroniske strukturer i det system, der studeres.
I eksperimenter, de "spektrale fingeraftryk" eller absorptionsspektret, måles med avancerede faciliteter såsom Advanced Light Source (ALS) ved det amerikanske energiministeriums Lawrence Berkeley National Laboratory (Berkeley Lab). I computersimuleringer, disse målinger er typisk fanget med en kvantemekanisk metode kaldet Time Dependent Density Functional Theory (TDDFT). Beregningsmodellerne er afgørende for at hjælpe forskere med at få mest muligt ud af deres eksperimenter ved at forudsige og validere resultater.
Men på trods af dets anvendelighed, der er tidspunkter, hvor TDDFT ikke kan bruges til at beregne absorptionsspektret for et system, fordi det ville kræve for meget tid og computerressourcer. Det er her, en ny matematisk "genvej" udviklet af forskere i Berkeley Labs Computational Research Division (CRD) kommer til nytte. Deres algoritme fremskynder absorptionsberegninger med en faktor på fem, så simuleringer, der plejede at tage 10 til 15 timer at beregne, kan nu udføres på cirka 2,5 timer.
Et papir, der beskriver denne metode, blev offentliggjort i Journal of Chemical Theory and Computation (JCTC). Og den nye tilgang til beregning af absorptionsspektret vil blive indarbejdet i en kommende udgivelse af den meget brugte NWChem computational chemistry software suite senere på året.
Nye algoritmer fører til beregningsbesparelser
At studere den kemiske struktur af nye molekyler og materialer, videnskabsmænd undersøger typisk systemet med en ekstern stimulus - typisk en laser - og leder derefter efter små elektroniske ændringer. Matematisk, denne elektroniske ændring kan udtrykkes som et egenværdiproblem. Ved at løse dette egenværdiproblem, forskere kan få en god tilnærmelse af absorptionsspektret, hvilket igen afslører resonansfrekvenserne for det system, der undersøges. I mellemtiden den tilsvarende egenvektor bruges til at beregne, hvor intenst systemet reagerede på stimulus. Dette er i bund og grund princippet bag TDDFT-tilgangen, som er blevet implementeret i adskillige kvantekemi-softwarepakker, inklusive open source NWChem-softwarepakken.
Selvom denne tilgang har vist sig at være vellykket, det har begrænsninger for store systemer. Jo bredere energiområdet af elektroniske svar en forsker forsøger at fange i et system, jo flere egenværdier og egenvektorer skal beregnes, hvilket også betyder, at flere computerressourcer er nødvendige. Ultimativt, absorptionsspektret af et molekylært system med mere end 100 atomer bliver uoverkommeligt dyrt at beregne med denne metode.
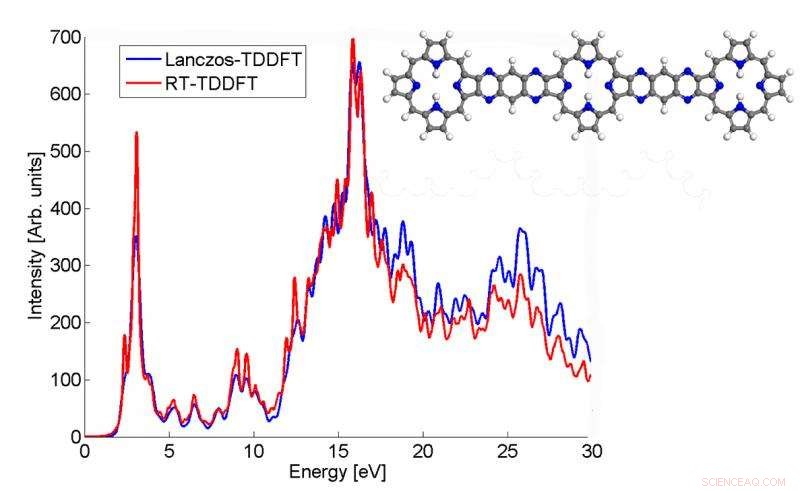
Dette plot viser, hvordan absorptionsspektret af et p3b2-molekyle beregnet af Lanczos-algoritmen stemmer overens med TDDFT-resultatet i realtid. Kredit:Chao Yang, Berkeley Lab
For at overvinde disse begrænsninger, matematikere i CRD udviklede en teknik til at beregne absorptionsspektret direkte uden eksplicit at beregne matricens egenværdier.
"Traditionelt forskere har været nødt til at beregne egenværdier og egenvektorer af meget store matricer for at generere absorptionsspektret, men vi indså, at du ikke behøver at beregne hver enkelt egenværdi for at få et præcist billede af absorptionsspektret, " siger Chao Yang, en CRD-matematiker, der ledede udviklingen af den nye tilgang.
Ved at omformulere problemet som en matrixfunktionstilnærmelse, at gøre brug af en særlig transformation og udnytte den underliggende symmetri med hensyn til en ikke-euklidisk metrisk, Yang og hans kolleger var i stand til at anvende Lanczos-algoritmen og en Kernal Polynomial Method (KPM) for at tilnærme absorptionsspektret af flere molekyler. Begge disse algoritmer kræver relativt lav hukommelse sammenlignet med ikke-symmetriske alternativer, som er nøglen til den beregningsmæssige besparelse.
Fordi denne metode kræver mindre computerkraft for at opnå et resultat, forskere kan også nemt beregne absorptionsspektret for molekylære systemer med flere hundrede atomer.
"Denne metode er et væsentligt skridt fremad, fordi den giver os mulighed for at modellere absorptionsspektret af molekylære systemer af hundredvis af atomer til lavere beregningsomkostninger." siger Niranjan Govind, en beregningskemiker ved Pacific Northwest National Laboratory, som samarbejdede med Berkeley Lab-teamet om udviklingen af metoden i NWChems computerkemiprogram.
For nylig brugte Berkeley Lab-forskere denne metode til at beregne absorptionsspektret og bekræfte, hvad adskillige eksperimentelle resultater har antydet - at grundstoffet berkelium bryder formen med sine tunge grundstoffers jævnaldrende ved at påtage sig en ekstra positiv ladning, når det er bundet til et syntetisk organisk molekyle. Denne egenskab kan hjælpe forskere med at udvikle bedre metoder til håndtering og rensning af nukleare materialer. Et papir, der fremhæver dette resultat, udkom den 10. april i tidsskriftet Naturkemi .
"De eksperimentelle resultater antydede denne usædvanlige adfærd i berkelium, men der var ikke nok eksperimentelle beviser til at sige ja, 100 procent, det er hvad vi ser, " siger studiemedforfatter Wibe Albert de Jong, en CRD-forsker. "For at være 100 procent sikker, vi lavede store beregningssimuleringer og sammenlignede dem med de eksperimentelle data og fandt ud af, at de var, Ja, ser berkelium i en usædvanlig oxidationstilstand."
Denne nye algoritme blev udviklet gennem et DOE Office of Science-støttet Scientific Discovery through Advanced Computing-projekt (SciDAC) fokuseret på at fremme software og algoritmer til fotokemiske reaktioner. SciDAC-projekter samler typisk et tværfagligt team af forskere for at udvikle nye og nye beregningsmetoder til at tackle nogle af de mest udfordrende videnskabelige problemer.
"Den tværfaglige karakter af SciDAC er en meget effektiv måde at lette banebrydende videnskab, da hvert teammedlem bringer et andet perspektiv til problemløsning, " siger Yang. "I dette dynamiske miljø, matematikere, ligesom mig, slå sig sammen med domæneforskere for at identificere beregningsmæssige flaskehalse, så bruger vi banebrydende matematiske teknikker til at adressere og overvinde disse udfordringer."
 Varme artikler
Varme artikler
-
 Fluorescerende sonder for at studere cellulær aktivitetFigur viser målretning af cPLA2 af den nydesignede inhibitor og substratsonde. Til venstre:Identificering af forskelle i cPLA2 -niveau i ubehandlet og Trichostatin A (TSA, en inhibitorforbindelse) -be
Fluorescerende sonder for at studere cellulær aktivitetFigur viser målretning af cPLA2 af den nydesignede inhibitor og substratsonde. Til venstre:Identificering af forskelle i cPLA2 -niveau i ubehandlet og Trichostatin A (TSA, en inhibitorforbindelse) -be -
 Forskere udvikler en ny raman-spektroskopiplatform til at karakterisere IDP'er i fortyndet opløsnin…En illustration, der viser det optiske pincet-kontrollerede hotspot til proteinstrukturkarakteriseringen ved overfladeforstærket Raman-spektroskopi. Kredit:Vince St. Dollente Mesias, Jinqing Huang / H
Forskere udvikler en ny raman-spektroskopiplatform til at karakterisere IDP'er i fortyndet opløsnin…En illustration, der viser det optiske pincet-kontrollerede hotspot til proteinstrukturkarakteriseringen ved overfladeforstærket Raman-spektroskopi. Kredit:Vince St. Dollente Mesias, Jinqing Huang / H -
 Ny metode til at opdage sygdomme, herunder coronavirus og cystisk fibroseIllustration viser ægteskabet mellem DNA -nanoteknologi og bioelektronik. Kredit:EatFishDesign En ny og hurtigere metode til diagnosticering af sygdomme hos patienter er blevet skabt af forskere v
Ny metode til at opdage sygdomme, herunder coronavirus og cystisk fibroseIllustration viser ægteskabet mellem DNA -nanoteknologi og bioelektronik. Kredit:EatFishDesign En ny og hurtigere metode til diagnosticering af sygdomme hos patienter er blevet skabt af forskere v -
 Solcreme og kosmetikforbindelser kan skade koraller ved at ændre fedtsyrerKredit:American Chemical Society Selvom solcreme er afgørende for at forebygge solskoldninger og hudkræft, nogle af dets ingredienser er ikke så gavnlige for havlevende skabninger. I særdeleshed,
Solcreme og kosmetikforbindelser kan skade koraller ved at ændre fedtsyrerKredit:American Chemical Society Selvom solcreme er afgørende for at forebygge solskoldninger og hudkræft, nogle af dets ingredienser er ikke så gavnlige for havlevende skabninger. I særdeleshed,
- Ekorre af Wisconsin
- USA's metan-emissioner er fladt siden 2006 på trods af øget olie- og gasaktivitet
- Somalisk præsident erklærer national katastrofe på grund af tørke
- Forskere ser COVID-19 som et historisk øjeblik for Storbritanniens miljømæssige fremtid
- Foreslået ingeniørmetode kunne bidrage til at gøre bygninger og broer sikrere
- Undersøgelse kortlægger by-landlige oplande og peger på måder at optimere politik- og planlægni…