


Kemikere lærer computerprogram til at modellere kræfter mellem atomer nøjagtigt

Kredit:MIPT
Et team af forskere fra MIPT, Skoltech, og Dukhov Research Institute of Automatics, ledet af Artem Oganov, brugte en machine learning -teknik til at modellere opførslen af aluminium og uran i de flydende og krystallinske faser ved forskellige temperaturer og tryk. Sådanne simuleringer af kemiske systemer kan forudsige deres egenskaber under en række betingelser, før eksperimenter udføres, muliggøre yderligere arbejde med kun de mest lovende materialer. Forskningsresultaterne blev offentliggjort i tidsskriftet Videnskabelige rapporter .
Computerkemi
Hurtige fremskridt inden for videnskaben i løbet af de sidste 100 år har resulteret i opdagelsen af et forbløffende antal organiske og uorganiske forbindelser, protein- og lipidstrukturer, og kemiske reaktioner. Men med alle disse nye strukturer og molekyler, en stigende tid er nødvendig for at studere deres makeup, biokemiske og fysiske egenskaber og at teste modellerne for deres adfærd under forskellige forhold og deres mulige interaktioner med andre forbindelser. Sådan forskning kan nu fremskyndes ved hjælp af computermodellering.
Kraftfeltmetoden er i øjeblikket den dominerende modelleringsteknik. Det gør brug af et sæt parametre, der beskriver et givet biokemisk system. Disse inkluderer bindingslængder, vinkler og ladninger, blandt andre. Imidlertid, denne teknik er ikke i stand til at gengive de kvantemekaniske kræfter, der spiller i molekyler, nøjagtigt. Nøjagtige kvantemekaniske beregninger er tidskrævende. Derudover de muliggør kun forudsigelser af adfærd for prøver, der i bedste fald er flere hundrede atomer store.
Maskinlæringsmetoder til molekylær modellering er af stor interesse for kemikere. De muliggør modeller, der er uddannet i relativt små datasæt opnået ved hjælp af kvantemekaniske beregninger. Sådanne modeller kan derefter erstatte kvantemekaniske beregninger, fordi de er lige præcise og kræver omkring 1, 000 gange mindre computerkraft.
Fremskridt med maskinlæringsværktøjer, der modellerer interaktioner mellem atomer
Forskerne brugte maskinlæring til at modellere interaktionerne mellem atomer i krystallinsk og flydende aluminium og uran. Aluminium er et velstuderet metal, hvis fysiske og kemiske egenskaber er kendt af forskere. Uran, derimod, blev valgt, fordi der er modstridende offentliggjorte data om dets fysiske og kemiske egenskaber, som forskerne søgte at definere mere præcist.
Papiret beskriver deres undersøgelse af sådanne materielle egenskaber som fonontætheden i tilstande, entropi, og smeltetemperaturen for aluminium.
"Størrelsen af interatomiske kræfter i krystaller kan bruges til at forudsige, hvordan atomer af det samme element vil opføre sig under forskellige temperaturer og i en anden fase, "siger Ivan Kruglov fra Computational Materials Design Laboratory på MIPT." Af samme grund, du kan bruge dataene om en væskes egenskaber til at finde ud af, hvordan atomerne vil opføre sig i en krystal. Det betyder, at ved at finde ud af mere om krystalstrukturen i uran, vi kan til sidst rekonstruere hele fasediagrammet for dette metal. Fasediagrammer er diagrammer, der angiver elementernes egenskaber som funktion af tryk og temperatur. De bruges til at bestemme grænserne for anvendelsen af et givet element. "
For at sikre, at data fra computersimuleringer er gyldige, de sammenlignes med eksperimentelle resultater. Metoden, som forskerne brugte, var i god overensstemmelse med tidligere forsøg. De oplysninger, der blev indhentet med metoden baseret på maskinlæring, havde en lavere fejlprocent, sammenlignet med modelleringsteknikkerne ved hjælp af kraftfelter.
I dette studie, forfatterne forbedrer deres 2016 -resultater med hensyn til hastigheden og nøjagtigheden af atomsystemmodellering ved hjælp af maskinlæring.
 Varme artikler
Varme artikler
-
 Forskellen mellem atomer, joner, molekyler og forbindelserEt enkelt sandkorn indeholder ca. 2,3 x 10 ^ 19 siliciumdioxidmolekyler. Det kan virke meget, men sandkornet indeholder endnu flere atomer end molekyler, da hvert siliciumdioxidmolekyle består af tre
Forskellen mellem atomer, joner, molekyler og forbindelserEt enkelt sandkorn indeholder ca. 2,3 x 10 ^ 19 siliciumdioxidmolekyler. Det kan virke meget, men sandkornet indeholder endnu flere atomer end molekyler, da hvert siliciumdioxidmolekyle består af tre -
 Ekstremt effektiv udvinding kan forbedre styringen af atombrændstofEn tetradentatligand vælger americium (Am, afbildet af grønne kugler) over europium (Eu, blå kugler). Rød angiver iltatomer og lilla, nitrogenatomer, der er nøglen til ligandens selektivitet. Kredit:O
Ekstremt effektiv udvinding kan forbedre styringen af atombrændstofEn tetradentatligand vælger americium (Am, afbildet af grønne kugler) over europium (Eu, blå kugler). Rød angiver iltatomer og lilla, nitrogenatomer, der er nøglen til ligandens selektivitet. Kredit:O -
 Computere deltager i kampen mod COVID-19Kredit:CC0 Public Domain Forskere overalt slår sig sammen for at bekæmpe den nye coronavirus, og de gør allerede fremskridt. Computation chemists er fokuseret på at bygge computermodeller af virus
Computere deltager i kampen mod COVID-19Kredit:CC0 Public Domain Forskere overalt slår sig sammen for at bekæmpe den nye coronavirus, og de gør allerede fremskridt. Computation chemists er fokuseret på at bygge computermodeller af virus -
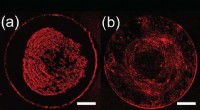 Ny fremstillingsteknik kan forbedre det almindelige problem inden for printteknologiFor figur A, der er partikeltransport til spidsen af måldråben, og der dannes en tæt midteraflejring. For figur B, den endelige kortlagte indbetaling er mere ensartet. Kredit: Langmuir En ny fr
Ny fremstillingsteknik kan forbedre det almindelige problem inden for printteknologiFor figur A, der er partikeltransport til spidsen af måldråben, og der dannes en tæt midteraflejring. For figur B, den endelige kortlagte indbetaling er mere ensartet. Kredit: Langmuir En ny fr
- Computermodel løser mysteriet om, hvordan gasbobler bygger store metanhydrataflejringer
- Bryder rekorden for optisk båndbredde for stabile pulserende lasere
- USA siger, at lækkende atomaffaldskuppel er sikker; Marshalløernes ledere tror ikke på det
- Cyber of the fittest:Forskere udvikler den første cyberagility-ramme til at måle angreb
- Ferskvand, der strømmer ind i det nordlige Stillehav, spiller en central rolle i klimaet i Nordamer…
- Forholdet mellem elektricitet og magnetisme