


Ny tilgang til løsning af proteinstrukturer fra små krystaller
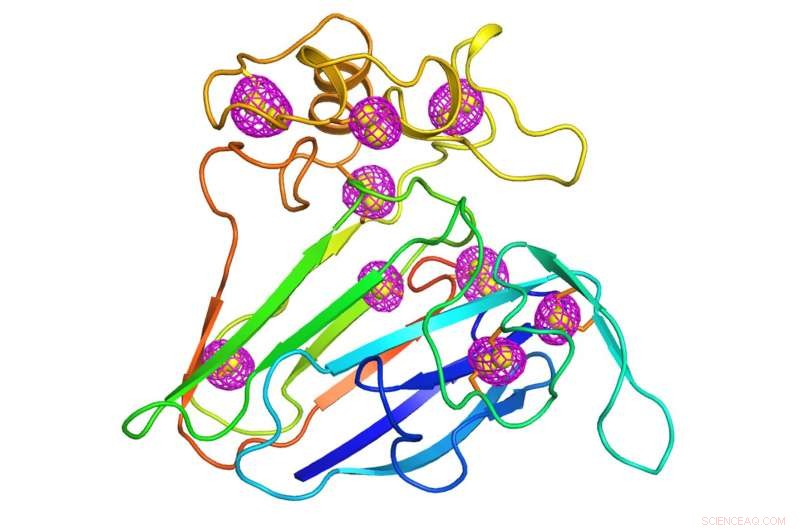
En tegneserie, der repræsenterer strukturen af et velstuderet planteprotein, der tjente som en testcase for den nyudviklede mikrokrystallografi teknik. Magenta maskemønstre, der omgiver svovlatomer, der er iboende for proteinet (gule kugler) angiver de uregelmæssige signaler, der blev ekstraheret ved hjælp af lavenergirøntgenstrålediffraktion af tusinder af krystaller, der måler mindre end 10 milliontedele af en meter, størrelsen af en bakterie. Kredit:Brookhaven National Laboratory
Brug af røntgenstråler til at afsløre atomskala 3-D strukturer af proteiner har ført til utallige fremskridt med at forstå, hvordan disse molekyler virker i bakterier, vira, planter, og mennesker - og har styret udviklingen af præcisionsmedicin til bekæmpelse af sygdomme som kræft og aids. Men mange proteiner kan ikke dyrkes til krystaller, der er store nok til, at deres atomarrangementer kan dechiffreres. For at tackle denne udfordring, forskere ved US Department of Energy's (DOE) Brookhaven National Laboratory og kolleger ved Columbia University har udviklet en ny tilgang til løsning af proteinstrukturer fra små krystaller.
Metoden er afhængig af unik prøvehåndtering, signaludtrækning, og tilgang til datamontering, og en strålelinje, der er i stand til at fokusere intense røntgenstråler ved Brookhaven's National Synchrotron Light Source II (NSLS-II)-en DOE Office of Science-brugerfacilitet-til et milliontedel-af-en-meter sted, omkring en halvtredsedel af et menneskehårs bredde.
"Vores teknik åbner virkelig døren for at håndtere mikrokrystaller, der tidligere har været utilgængelige, herunder vanskeligt krystalliserede celleoverfladereceptorer og andre membranproteiner, fleksible proteiner, og mange komplekse humane proteiner, "sagde Brookhaven Labs videnskabsmand Qun Liu, den tilsvarende forfatter til undersøgelsen, som blev offentliggjort den 3. maj, 2019, i IUCrJ , et tidsskrift for International Union of Crystallography.
Dekryptering af proteinstrukturer
Proteinkrystallografi har været en dominerende metode til løsning af proteinstrukturer siden 1958, forbedres med tiden, da røntgenkilder er blevet mere kraftfulde, muliggøre mere præcise strukturbestemmelser. For at bestemme en proteinstruktur, forskere måler, hvordan røntgenstråler som dem, der genereres ved NSLS-II, afviger, eller hoppe af, atomerne i et ordnet krystallinsk gitter bestående af mange kopier af det samme proteinmolekyle, der alle var anbragt på samme måde. Diffraktionsmønsteret formidler information om, hvor atomerne er placeret. Men det er ikke tilstrækkeligt.
"Kun amplituderne af diffrakterede røntgenbølger" registreres på detektoren, men ikke deres faser (timingen mellem bølger), "sagde Liu." Begge er nødvendige for at rekonstruere en 3D-struktur. Dette er det såkaldte krystallografiske faseproblem. "
Krystallografer har løst dette problem ved at indsamle fasedata fra en anden form for spredning, kendt som anomal spredning. Anomal spredning opstår, når atomer er tungere end et proteins hovedbestanddele af kulstof, brint, og nitrogen absorberer og udsender nogle af røntgenstrålerne igen. Dette sker, når røntgenenergien er tæt på den energi, som tunge atomer kan lide at absorbere. Forskere indsætter undertiden kunstigt tunge atomer såsom selen eller platin i proteinet til dette formål. Men svovlatomer, der forekommer naturligt i proteinmolekyler, kan også producere sådanne signaler, omend svagere. Selvom disse afvigende signaler er svage, en stor krystal har normalt nok kopier af proteinet med nok svovlatomer til at gøre dem målbare. Det giver forskere den faseinformation, der er nødvendig for at lokalisere svovlatomernes placering og oversætte diffraktionsmønstrene til en fuld 3D-struktur.
"Når du kender svovlpositionerne, du kan beregne faserne for de andre proteinatomer, fordi forholdet mellem svovl og de andre atomer er fast, "sagde Liu.
Men små krystaller, Per definition, ikke har så mange kopier af proteinet af interesse. Så i stedet for at lede efter diffraktions- og faseinformation fra gentagne kopier af et protein i en enkelt stor krystal, Brookhaven/Columbia -teamet udviklede en måde at tage målinger fra mange små krystaller, og derefter samle de kollektive data.
Små krystaller, store resultater
For at håndtere de små krystaller, teamet udviklede prøvegitter mønstret med brønde i mikrostørrelse. Efter hældning af opløsningsmiddel indeholdende mikrokrystallerne over disse godt monterede gitre, forskerne fjernede opløsningsmidlet og frøs de krystaller, der var fanget på netene.
"Vi har stadig en udfordring, selvom, fordi vi ikke kan se, hvor de små krystaller er på vores gitter, "sagde Liu." For at finde ud af det, vi brugte mikrodiffraktion ved NSLS-II's Frontier Microfocusing Macromolecular Crystallography (FMX) beamline til at undersøge hele nettet. Scanning linje for linje, vi kan finde, hvor disse krystaller er skjult. "
Som Martin Fuchs, den ledende stråleforsker ved FMX, forklaret, "FMX beamline kan fokusere den fulde intensitet af røntgenstrålen ned til en størrelse på en mikron, eller milliontedel af en meter. Vi kan fint styre strålestørrelsen for at matche den til størrelsen af krystallerne - fem mikron i tilfælde af det nuværende eksperiment. Disse muligheder er afgørende for at opnå det bedste signal, " han sagde.
Wuxian Shi, en anden FMX beamline -forsker, bemærkede, at "de data, der er indsamlet i gitterundersøgelsen, indeholder oplysninger om krystallernes placering. Derudover kan vi kan også se, hvor godt hver krystal diffrakterer, som tillader os kun at vælge de bedste krystaller til dataindsamling. "
Forskerne var derefter i stand til at manøvrere prøveholderen for at placere hver kortlagt mikrokrystal af interesse tilbage i midten af præcisionsrøntgenstrålen til dataindsamling.
De brugte den laveste energi til rådighed ved strålelinjen - indstillet til at nærme sig så tæt som muligt svovlatomers absorptionsenergi - og indsamlede unormale spredningsdata.
"De fleste krystallografiske strålelinjer kunne ikke nå svovlabsorptionskanten for optimerede afvigende signaler, "sagde medforfatter Wayne Hendrickson fra Columbia University." Heldigvis NSLS-II er en verdensførende synkrotron lyskilde, der leverer lyse røntgenstråler, der dækker et bredt spektrum af røntgenenergi. Og selvom vores energiniveau var lidt over den ideelle absorptionsenergi for svovl, det genererede de uregelmæssige signaler, vi havde brug for. "
Men forskerne havde stadig noget at gøre for at udtrække de vigtige signaler og samle data fra mange små krystaller.
"Vi får faktisk tusindvis af data, "sagde Liu." Vi brugte omkring 1400 mikrokrystaller, hver med sit eget datasæt. Vi er nødt til at sammensætte alle data fra disse mikrokrystaller. "
They also had to weed out data from crystals that were damaged by the intense x-rays or had slight variations in atomic arrangements.
"A single microcrystal does not diffract x-rays sufficiently for structure solution prior to being damaged by the x-rays, " said Sean McSweeney, deputy photon division director and program manager of the Structural Biology Program at NSLS-II. "This is particularly true with crystals of only a few microns, the size of about a bacterial cell. We needed a way to account for that damage and crystal structure variability so it wouldn't skew our results."
They accomplished these goals with a sophisticated multi-step workflow process that sifted through the data, discarded outliers that might have been caused by radiation damage or incompatible crystals, and ultimately extracted the anomalous scattering signals.
"This is a critical step, " said Liu. "We developed a computing procedure to assure that only compatible data were merged in a way to align the individual microcrystals from diffraction patterns. That gave us the required signal-to-noise ratios for structure determination."
Applying the technique
This technique can be used to determine the structure of any protein that has proven hard to crystallize to a large size. These include cell-surface receptors that allow cells of advanced lifeforms such as animals and plants to sense and respond to the environment around them by releasing hormones, transmitting nerve signals, or secreting compounds associated with cell growth and immunity.
"To adapt to the environment through evolution, these proteins are malleable and have lots of non-uniform modifications, " said Liu. "It's hard to get a lot of repeat copies in a crystal because they don't pack well."
Hos mennesker, receptors are common targets for drugs, so having knowledge of their varied structures could help guide the development of new, more targeted pharmaceuticals.
But the technique is not restricted to just small crystals.
"The method we developed can handle small protein crystals, but it can also be used for any size protein crystals, any time you need to combine data from more than one sample, "Sagde Liu.
 Varme artikler
Varme artikler
-
 Antiferromagnetiske materialer kæmper frem mod kommerciel anvendelseFig.1:Et skematisk diagram af informationslagring ved brug af konventionelle ferromagnet (FM)-baserede spintroniske enheder (venstre) og de foreslåede antiferromagneter (AFMer)-baserede enheder (højre
Antiferromagnetiske materialer kæmper frem mod kommerciel anvendelseFig.1:Et skematisk diagram af informationslagring ved brug af konventionelle ferromagnet (FM)-baserede spintroniske enheder (venstre) og de foreslåede antiferromagneter (AFMer)-baserede enheder (højre -
 Hvad laver calciumchlorid & bagesoda?Kombination af calciumchlorid og bagepulver - natriumbicarbonat - i en forseglelig plastikpose er et foretrukket kemisk eksperiment på gymnasiet. Det producerer en gas, så hvis du forsegler posen e
Hvad laver calciumchlorid & bagesoda?Kombination af calciumchlorid og bagepulver - natriumbicarbonat - i en forseglelig plastikpose er et foretrukket kemisk eksperiment på gymnasiet. Det producerer en gas, så hvis du forsegler posen e -
 Programmering af DNA til at levere kræftmedicinDNA har en vigtig opgave - det fortæller dine celler, hvilke proteiner de skal lave. Nu, et forskerhold ved University of Delaware har udviklet teknologi til at programmere DNA-strenge til switche, de
Programmering af DNA til at levere kræftmedicinDNA har en vigtig opgave - det fortæller dine celler, hvilke proteiner de skal lave. Nu, et forskerhold ved University of Delaware har udviklet teknologi til at programmere DNA-strenge til switche, de -
 Vi skabte diamanter på få minutter uden varme ved at efterligne kraften ved en asteroide -kollisio…Krystalstrukturerne i kubisk diamant og sekskantet Lonsdaleite har atomer arrangeret forskelligt. I naturen, diamanter dannes dybt i jorden over milliarder af år. Denne proces kræver miljøer med u
Vi skabte diamanter på få minutter uden varme ved at efterligne kraften ved en asteroide -kollisio…Krystalstrukturerne i kubisk diamant og sekskantet Lonsdaleite har atomer arrangeret forskelligt. I naturen, diamanter dannes dybt i jorden over milliarder af år. Denne proces kræver miljøer med u
- Forskere opdager årsagen til atlanterhavskystens stigning i havniveauet hot spots
- De 3 typer bakterier
- Første direkte observation af orienteret vedhæftning i nanokrystalvækst
- Hvordan er bevægelsesmåling?
- Hvordan et boreskib trækker kerner fra 4 km under havet
- Forskellen mellem en børstet og børsteløs motor