


Forskningsgruppen bruger supercomputing til at målrette de mest lovende lægemiddelkandidater ud fra et frygtindgydende antal muligheder
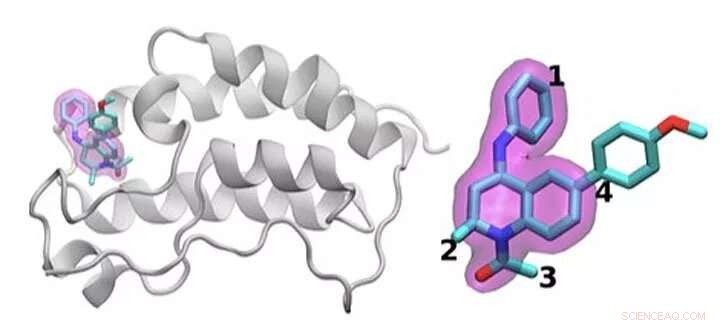
En skematisk oversigt over BRD4 -proteinet bundet til et af 16 lægemidler baseret på det samme tetrahydroquinolin -stillads (fremhævet i magenta). Regioner, der er kemisk modificeret mellem de lægemidler, der undersøges i denne undersøgelse, er mærket 1 til 4. Typisk der foretages kun en lille ændring af den kemiske struktur fra et lægemiddel til det næste. Denne konservative tilgang gør det muligt for forskere at undersøge, hvorfor et lægemiddel er effektivt, mens et andet ikke er det. Kredit:Brookhaven National Laboratory
At identificere den optimale lægemiddelbehandling er som at ramme et bevægeligt mål. For at stoppe sygdom, småmolekylære lægemidler binder tæt til et vigtigt protein, blokerer dens virkninger i kroppen. Selv godkendte lægemidler virker normalt ikke hos alle patienter. Og med tiden, infektiøse midler eller kræftceller kan mutere, gør et engang effektivt stof ubrugeligt.
Et kerne -fysisk problem ligger til grund for alle disse spørgsmål:optimering af interaktionen mellem lægemiddelmolekylet og dets proteinmål. Variationerne i lægemiddelkandidatmolekyler, mutationsområdet i proteiner og den overordnede kompleksitet af disse fysiske interaktioner gør dette arbejde svært.
Shantenu Jha fra Department of Energy's (DOE's) Brookhaven National Laboratory og Rutgers University leder et team, der forsøger at strømline beregningsmetoder, så supercomputere kan påtage sig en del af denne enorme arbejdsbyrde. De har fundet en ny strategi for at tackle en del:at differentiere, hvordan lægemiddelkandidater interagerer og binder med et målrettet protein.
For deres arbejde, Jha og hans kolleger vandt sidste års IEEE International Scalable Computing Challenge (SCALE) -pris, som genkender skalerbare databehandlingsløsninger til videnskabelige og tekniske problemer i den virkelige verden.
For at designe et nyt lægemiddel, et farmaceutisk selskab kan starte med et bibliotek med millioner af kandidatmolekyler, som de indsnævrer til de tusinder, der viser en indledende binding til et målprotein. At forfine disse muligheder til et nyttigt lægemiddel, der kan testes hos mennesker, kan involvere omfattende eksperimenter for at tilføje eller fratrække atomgrupper på vigtige steder på molekylet og teste, hvordan hver af disse ændringer ændrer, hvordan det lille molekyle og protein interagerer.
Simuleringer kan hjælpe med denne proces. Større, hurtigere supercomputere og stadig mere sofistikerede algoritmer kan inkorporere realistisk fysik og beregne bindingsenergierne mellem forskellige små molekyler og proteiner. Sådanne metoder kan forbruge betydelige beregningsressourcer, imidlertid, for at opnå den nødvendige nøjagtighed. Industri-nyttige simuleringer skal også give hurtige svar. På grund af tovtrækkeriet mellem nøjagtighed og hastighed, forskere fornyer konstant, udvikling af mere effektive algoritmer og forbedring af ydeevnen, Siger Jha.
Dette problem kræver også at styre beregningsressourcer anderledes end for mange andre store problemer. I stedet for at designe en enkelt simulering, der skalerer til at bruge en hel supercomputer, forskere kører samtidigt mange mindre modeller, der former hinanden og banen for fremtidige beregninger, en strategi kendt som ensemblebaseret computing, eller komplekse arbejdsgange.
"Tænk på dette som at prøve at udforske et meget stort åbent landskab for at forsøge at finde ud af, hvor du måske kan få den bedste lægemiddelkandidat, "Siger Jha. Tidligere har forskere har bedt computere om at navigere i dette landskab ved at foretage tilfældige statistiske valg. På et beslutningssted, halvdelen af beregningerne kan følge en vej, den anden halvdel en anden.
Jha og hans team søger måder at hjælpe disse simuleringer på at lære af landskabet i stedet. Indtagelse og derefter deling af data i realtid er ikke let, Jha siger, "og det var det, der krævede noget af den teknologiske innovation at gøre i stor skala." Han og hans Rutgers-baserede team samarbejder med Peter Coveney's gruppe ved University College London om dette arbejde.
For at teste denne idé, de har brugt algoritmer, der forudsiger bindingsaffinitet og har introduceret strømlinede versioner i en HTBAC -ramme, til bindingsaffinitetsberegner med høj gennemstrømning. En sådan lommeregner, kendt som ESMACS, hjælper dem med at fjerne molekyler, der binder dårligt til et målprotein. Den anden, Bånd, er mere præcis, men mere begrænset i omfang og kræver 2,5 gange flere beregningsressourcer. Ikke desto mindre, det kan hjælpe forskerne med at optimere en lovende interaktion mellem et lægemiddel og et protein. HTBAC -rammen hjælper dem med at implementere disse algoritmer effektivt, gemme den mere intensive algoritme til situationer, hvor det er nødvendigt.
Teamet demonstrerede ideen ved at undersøge 16 lægemiddelkandidater fra et molekylbibliotek på GlaxoSmithKline (GSK) med deres mål, BRD4-BD1-et protein, der er vigtigt i brystkræft og inflammatoriske sygdomme. Lægemiddelkandidaterne havde den samme kernestruktur, men adskilte sig på fire forskellige områder omkring molekylets kanter.
I denne indledende undersøgelse kørte teamet tusinder af processer samtidigt på 32, 000 kerner på Blue Waters, en supercomputer fra National Science Foundation (NSF) ved University of Illinois i Urbana-Champaign. De har kørt lignende beregninger på Titan, supercomputer Cray XK7 på Oak Ridge Leadership Computing Facility, en DOE Office of Science brugerfacilitet. Teamet skelnede med succes mellem bindingen af disse 16 lægemiddelkandidater, den hidtil største sådan simulering. "Vi nåede ikke bare en hidtil uset skala, "Jha siger." Vores tilgang viser evnen til at differentiere. "
De vandt deres SCALE -pris for dette første bevis på koncept. Udfordringen nu, Jha siger, sørger for, at det ikke kun virker for BRD4, men også for andre kombinationer af lægemiddelmolekyler og proteindivider.
Hvis forskerne kan fortsætte med at udvide deres tilgang, sådanne teknikker kan i sidste ende hjælpe med at fremskynde opdagelse af lægemidler og muliggøre personlig medicin. Men for at undersøge mere realistiske problemer, de får brug for mere beregningskraft. "Vi er midt i denne spænding mellem et meget stort kemisk rum, som vi, i princippet, har brug for at udforske, og, desværre begrænsede computerressourcer. "siger Jha.
Selvom supercomputing ekspanderer mod exascale, beregningsforskere kan mere end udfylde hullet ved at tilføje mere realistisk fysik til deres modeller. I en overskuelig fremtid, forskere skal være ressourcestærke for at skalere disse beregninger. Nødvendighed er moder til innovation, Jha siger, netop fordi molekylær videnskab ikke vil have den ideelle mængde beregningsressourcer til at udføre simuleringer.
Men exascale computing kan hjælpe med at flytte dem tættere på deres mål. Udover at arbejde med University College London og GSK, Jha og hans kolleger samarbejder med Rick Stevens fra Argonne National Laboratory og CANcer Distributed Learning Environment (CANDLE) -teamet. Dette co-design projekt inden for DOE's Exascale Computing Project bygger dybe neurale netværk og generelle maskinlæringsteknikker til undersøgelse af kræft. Algoritmerne og softwaren inden for HTBAC kunne supplere CANDLEs fokus på disse tilgange.
Dette bredere samarbejde mellem Jhas gruppe, CANDLE-teamet og John Choderas laboratorium ved Memorial Sloan-Kettering Cancer Center har ført til projektet Integrated and Scalable Prediction of Resistance (INSPIRE). Dette team har allerede kørt simuleringer på DOE's Summit -supercomputer på Oak Ridge National Laboratory. Det vil snart fortsætte dette arbejde med Frontera - NSF's ledermaskine ved University of Texas i Austins Texas Advanced Computing Center.
"Vi er sultne efter større fremskridt og større metodiske forbedringer, "Jha siger." Vi vil gerne se, hvordan disse smukke komplementære tilgange integrativt kan arbejde hen imod denne storslåede vision. "
 Varme artikler
Varme artikler
-
 Forskere udvikler DNA-solcreme, der bliver bedre, jo længere du bruger denEn afbildning af den dobbelte spiralformede struktur af DNA. Dens fire kodningsenheder (A, T, C, G) er farvekodet i pink, orange, lilla og gul. Kredit:NHGRI Hvorfor bruge almindelig solcreme, når
Forskere udvikler DNA-solcreme, der bliver bedre, jo længere du bruger denEn afbildning af den dobbelte spiralformede struktur af DNA. Dens fire kodningsenheder (A, T, C, G) er farvekodet i pink, orange, lilla og gul. Kredit:NHGRI Hvorfor bruge almindelig solcreme, når -
 Registrering af forurening med en kompakt laserkildeKredit:Ecole Polytechnique Federale de Lausanne (EPFL) Forskere ved EPFL er kommet med en ny mellem infrarød lyskilde, der kan registrere drivhusgasser og andre gasser, samt molekyler i en persons
Registrering af forurening med en kompakt laserkildeKredit:Ecole Polytechnique Federale de Lausanne (EPFL) Forskere ved EPFL er kommet med en ny mellem infrarød lyskilde, der kan registrere drivhusgasser og andre gasser, samt molekyler i en persons -
 Optrævling af den molekylære kompleksitet af cellulære maskiner og miljøprocesser21-Tesla Fourier transformation ion cyclotron resonans massespektrometer (data vist til højre) vil drive den fremtidige retning af miljø, biologiske, atmosfærisk, og energiforskning. Kredit:Pacific No
Optrævling af den molekylære kompleksitet af cellulære maskiner og miljøprocesser21-Tesla Fourier transformation ion cyclotron resonans massespektrometer (data vist til højre) vil drive den fremtidige retning af miljø, biologiske, atmosfærisk, og energiforskning. Kredit:Pacific No -
 Hvad skrives nummeret til venstre for det kemiske symbol eller den formel, der kaldes?Den eneste gang du ser et tal til venstre for formlen for en kemisk forbindelse er, når forbindelsen er involveret i en reaktion, og du ser på ligningen for reaktionen. Når du ser et tal i denne sa
Hvad skrives nummeret til venstre for det kemiske symbol eller den formel, der kaldes?Den eneste gang du ser et tal til venstre for formlen for en kemisk forbindelse er, når forbindelsen er involveret i en reaktion, og du ser på ligningen for reaktionen. Når du ser et tal i denne sa
- Ny enhed kan gøre konvertering af spildvarme til elektricitet industrielt konkurrencedygtig
- Ideer til et CO2-bilprojekt
- Forskere udvikler hårdere keramik til panservinduer
- Kaster lys på den mørke side af biomassebrændende forurening
- Statistikker over ekstrem nedbør kan ændre sig, efterhånden som det amerikanske klima opvarmes
- The Plants & Animals of the Coastal Plain