


En vanskelig reaktionssekvens får et stort løft fra et flow-opsætning og statistik
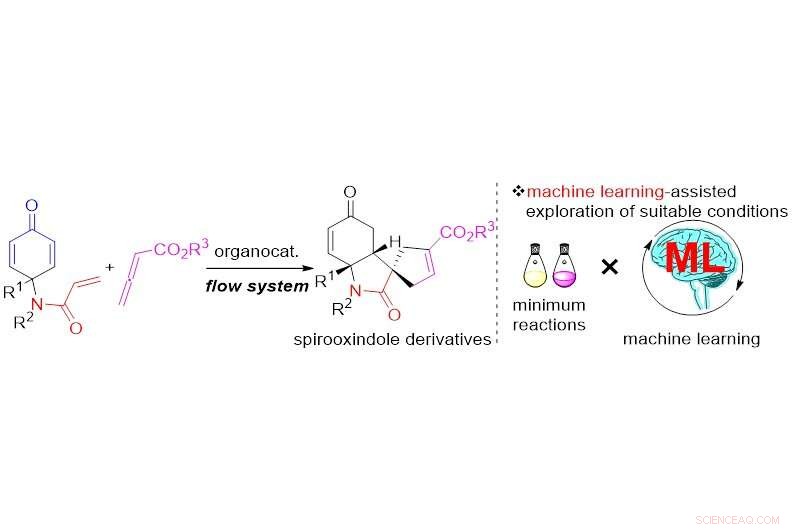
Maskinstøttet udforskning af organokatalyseret dominoreaktion i flowsystem. Kredit:Royal Society of Chemistry
Forskere fra Osaka University optimerer en kompliceret dominoreaktion i et flowsystem via maskinlæring for effektivt at screene flere variabler, opnå høj selektivitet og udbytte af en potentiel biologisk aktiv forbindelse
På trods af teknologiske fremskridt, tidlig opdagelse og udvikling af lægemidler er stadig tidskrævende, vanskelig og ineffektiv proces med lave succesrater. Et team fra Osaka University har opdaget en mulig løsning til at overvinde lave produktionsudbytter i komplekse reaktionssekvenser, tilvejebringelse af en proof-of-concept undersøgelse af det vellykkede høje udbytte af et potentielt terapeutisk middel.
I en undersøgelse for nylig offentliggjort i Kemisk kommunikation , forskerne demonstrerer produktionen af et potentielt lægemiddel ved hjælp af maskinlæring til hurtigt at screene eksperimentelle forhold for en kompleks reaktionsserie. Denne optimeringsmetode reducerede tiden betydeligt, materialer og omkostninger, der kræves til konventionelle metoder.
For både akademiske og industrielle forskere, et væsentligt skridt i udviklingen af kemiske reaktioner involverer optimering af eksperimentelle forhold. Dette opnås traditionelt ved at variere én parameter og holde de andre konstante - en besværlig og omkostningsfuld proces. En strategi til hurtigt at identificere optimale parametre er maskinlæring, et statistisk værktøj, der bruges på mange områder, herunder opdagelse af lægemidler.
"Mens jeg undersøgte trinene i den organokatalyserede Rauhut – Currier og [3+2] annuleringssekvens, vi indså først, at et mikroblandingsstrømsystem ville undertrykke eventuelle uønskede sidereaktioner og forbedre udbyttet af det ønskede biologisk aktive spirooxindolderivat, "siger seniorforfatter af undersøgelsen, Hiroaki Sasai. "Den Gaussiske procesregression (GPR) gav os derefter mulighed for hurtigt at screene forskellige parametre og udforske de optimale flowbetingelser for vores system for at maksimere produktudbyttet."
Disse spirooxindol motiver, findes i mange biologisk aktive molekyler og naturlige produkter, har vundet betydelig forskningsinteresse som mulige antivirale lægemidler. Som med andre stoffer, fremstilling af spirooxindoler resulterer i blandinger, der indeholder spejlbilledevarianter af det samme molekyle (enantiomerer) med forskellige kemiske egenskaber (f.eks. lægemiddelaktivitet vs. ingen aktivitet) - den vanskelige del er fortrinsvis at maksimere udbyttet af den ønskede variant, der viser lægemiddelaktivitet. En forenklet metode til at opnå denne bedrift med spirooxindoler har indtil nu stort set været uden for rækkevidde.
På trods af kompleksiteten, selektivitet og specificitet af den højeffektive reaktionssekvens, forskerne etablerede reaktionen ved hjælp af et mikro-mixer flow system, omend med 49% udbytte. Ved at bruge de optimerede parametre fra GPR, de opnåede derefter spirooxindolderivaterne med tre sammenhængende chirale centre inden for et minut med op til 89% udbytte og 98% renhed af den ønskede spejlbilledvariant.
"Det er udfordrende at forudsige effekten af at ændre hver eksperimentel parameter, når man udvikler en ny reaktion uden en grundig reaktionsoptimering, " forklarer hovedforfatter Masaru Kondo. "Men, at kombinere værktøjer som GPR med nye syntetiske metoder i flowsystemer kan forenkle og strømline lægemiddeludviklingsprocessen for andre komplicerede molekyler, reducere omkostninger, tid og materialespild."
Sidste artikelEt stof, mange sygdomme
Næste artikelRevner gør historiske malerier mindre sårbare over for miljømæssige variationer
 Varme artikler
Varme artikler
-
 Lysdrevne nano-organismer forbruger CO2, skabe miljøvenlig plast og brændstofUniversity of Colorado Boulder Adjunkt Prashant Nagpal Kredit:Casey A. Cass University of Colorado Boulder forskere har udviklet nanobio-hybride organismer, der er i stand til at bruge luftbåren k
Lysdrevne nano-organismer forbruger CO2, skabe miljøvenlig plast og brændstofUniversity of Colorado Boulder Adjunkt Prashant Nagpal Kredit:Casey A. Cass University of Colorado Boulder forskere har udviklet nanobio-hybride organismer, der er i stand til at bruge luftbåren k -
 Smart blæk tilføjer nye dimensioner til 3D-udskrivningEt eksempel fra forskningen viser, hvordan et 3D-printet objekt sammensat af hydrogel (G1) kan ændre størrelse efter udskrivning. Selvom dette eksempel tjener til at demonstrere resultatet, andre obje
Smart blæk tilføjer nye dimensioner til 3D-udskrivningEt eksempel fra forskningen viser, hvordan et 3D-printet objekt sammensat af hydrogel (G1) kan ændre størrelse efter udskrivning. Selvom dette eksempel tjener til at demonstrere resultatet, andre obje -
 Forskere udvikler ny teknik til produktion af plasmonics-enhederKredit:CC0 Public Domain Forskningslaboratorier udvikler konstant nye materialer, der forventes at udvise nye egenskaber, der er bundet til at revolutionere denne eller hin teknologi. Men det er i
Forskere udvikler ny teknik til produktion af plasmonics-enhederKredit:CC0 Public Domain Forskningslaboratorier udvikler konstant nye materialer, der forventes at udvise nye egenskaber, der er bundet til at revolutionere denne eller hin teknologi. Men det er i -
 Forskere syntetiserer vedvarende olier til brug i smøremidlerKredit:CC0 Public Domain Motor gear, fly thrustere, køleskabskompressorer, vindmøller - listen over vigtige industrimaskiner, landbrugsudstyr, transportfartøjer, og hjemmeapplikationer, der er afh
Forskere syntetiserer vedvarende olier til brug i smøremidlerKredit:CC0 Public Domain Motor gear, fly thrustere, køleskabskompressorer, vindmøller - listen over vigtige industrimaskiner, landbrugsudstyr, transportfartøjer, og hjemmeapplikationer, der er afh
- NOAA-satellitter hjalp med at redde 307 liv i 2016
- Virus bringer luftfartens fremtid i fare uden statsstøtte:Lufthansa
- Undersøgelse identificerer sandsynlige scenarier for global spredning af ødelæggende afgrødesygd…
- Snapchat løfter sløret for nye originale shows under eget brand
- Menneskerettighedstraktater gavner verdens mest undertrykte
- Genbrug af ansigtsmasker på veje for at håndtere COVID-genereret affald