


Forudsigelse af røntgenabsorptionsspektre fra grafer
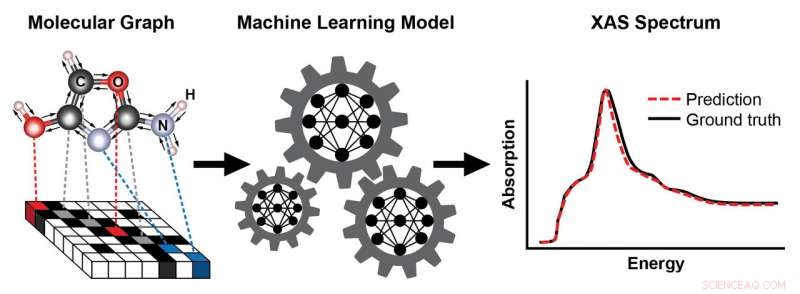
Et skema, der viser trinene til træning af en maskinlæringsmodel til at forudsige et røntgenabsorptionsspektrum (XAS) baseret på den kendte struktur af et molekyle. Molekylets struktur er repræsenteret som en graf, med atomer som noder og kemiske bindinger som kanter. Denne repræsentation fanger atomernes forbindelse – her, kulstof (C), oxygen (O), nitrogen (N), og hydrogen (H) - og typen og længden af de kemiske bindinger, der forbinder dem. Det resulterende XAS-spektrum indeholder rig information om det lokale kemiske miljø af absorberende atomer, såsom deres symmetri og antallet af naboatomer. Kredit:Brookhaven National Laboratory
Røntgenabsorptionsspektroskopi (XAS) er en populær karakteriseringsteknik til at undersøge den lokale atomstruktur og elektroniske egenskaber af materialer og molekyler. Fordi atomer af hvert element absorberer røntgenstråler ved karakteristiske energier, XAS er velegnet til at kortlægge den rumlige fordeling af elementer i en prøve. Typisk, forskere udfører XAS-eksperimenter ved synkrotronlyskilder - såsom National Synchrotron Light Source II (NSLS-II) - fordi de giver meget lys, afstembare røntgenstråler. Ved at måle absorbansen i en prøve ved varierende røntgenenergier, forskere kan generere et plot kaldet et røntgenabsorptionsspektrum.
"XAS er en nøglefunktion for brugere på Brookhaven National Laboratory's NSLS-II og Center for Functional Nanomaterials (CFN), både US Department of Energy (DOE) Office of Science brugerfaciliteter, der er åbne for det videnskabelige forskningssamfund, " sagde Deyu Lu, en fysiker i CFN Theory and Computation Group. "Med de rigtige analyseværktøjer, XAS kan give en enorm indsigt i nanovidenskabelig forskning. Udviklingen af sådanne værktøjer er central for vores mission som brugerfaciliteter."
Klassificering af lokale kemiske miljøer
Forskellige områder af røntgenabsorptionsspektret er følsomme over for forskellige aspekter af materialeegenskaberne i en prøve. For eksempel, X-ray absorption near-edge structure (XANES) fokuserer på spektrets nærkantsområde, lige over startenergien tilstrækkelig til at excitere en elektron fra et atoms indre skaller til en tom tilstand. XANES koder rig information om det lokale kemiske miljø af absorberende atomer i en prøve - inklusive deres geometriske koordination, symmetri, og ladningstilstand (antallet af elektroner opnået eller tabt fra kemisk binding). Men at analysere spektrale data er meget udfordrende på grund af deres abstrakte natur.
"I modsætning til et mikroskopbillede af et materiale, hvor du direkte kan se funktioner som krystallinitet eller defekter, XANES spektre koder information, der kræver domæneekspertise at fortolke, " forklarede Lu.
Standardfortolkning af signaler i et XANES-spektrum er afhængig af karakteristiske træk kendt som "fingeraftryk, " som er konstrueret ud fra målinger på referencematerialer. denne fingeraftrykstilgang mislykkes, når prøven ikke er en simpel krystal, og relevante referencematerialer ikke let kan identificeres.
Storskala teoribaserede simuleringer fra atomstrukturmodeller kan give meget nyttig indsigt til fortolkningen af eksperimentelle XANES-spektre; imidlertid, disse simuleringer er ofte beregningsmæssigt dyre og tidskrævende, og deres nøjagtighedsniveau afhænger i høj grad af de valgte teoretiske tilnærmelser og det undersøgte system. Som resultat, robust spektral fortolkning er i øjeblikket flaskehalsen i XAS-studier. Desuden, Realtidsfortolkning af XAS-spektre er dukket op som en ny udfordring for studier af den dynamiske udvikling af materialer under driftsforhold og autonome eksperimenter. Behovet for robuste, effektiv spektral fortolkning bliver stadig mere udbredt ved synkrotronlyskilder.
"Realtid, nøjagtig fortolkning af røntgenspredning og spektroskopimålinger såsom røntgenabsorption, fluorescens, og diffraktion er en vigtig egenskab for brugere, der udfører forskning på NSLS-II og andre synkrotronlysanlæg, " sagde Mehmet Topsakal, en videnskabelig medarbejder i Materials for Energy Applications Group i Brookhavens Nuclear Science and Technology Department, som udvikler avanceret dataanalyse og maskinlæringsteknikker til røntgenspektroskopi. "Hvert år, tusindvis af forskere fra hele verden kommer til NSLS-II for at undersøge egenskaberne af forskellige materialer. En state-of-the-art spektralanalysepipeline ville give brugerne mulighed for at få nyttig feedback på deres prøver, mens eksperimenter er i gang, og foretage justeringer i farten for at guide eksperimenter. Spørgsmålet er, hvordan kan vi udføre spektral fortolkning i realtid for at afdække struktur-spektrum korrelationer?"
Udtrække information med maskinlæring
Udnyttelse af big data og machine learning, Lu og Topsakal satte sig for at besvare dette spørgsmål med beregningsforsker Shinjae Yoo fra Brookhaven Labs Computational Science Initiative (CSI) og Columbia University Ph.D. kandidat og DOE Computational Science Graduate Fellow Matthew Carbone.
"DOE Computational Science Graduate Fellowship har givet mig en unik mulighed for at strække mig ud over min ph.d.-forskning i kemisk fysik ved Columbia for at udforske kraften ved maskinlæringsalgoritmer, arbejder sammen med Brookhaven-forskere, " sagde Carbone. "Maskinlæring udnytter massive datasæt til at bygge meget indsigtsfulde modeller, der en gang trænet, kan lave forudsigelser på farten på nye data. Sådanne modeller kunne bruges til at omgå dyre kvantekemiberegninger og støtte i operando-materialekarakterisering."
Medlemmer af dette team og samarbejdspartnere har arbejdet på spektrum-til-struktur- og struktur-til-spektrum-kortlægninger i flere år. I 2017 de udviklede maskinlæringsmodeller til at forudsige det gennemsnitlige koordinationstal for metalnanopartikler fra XANES-spektre. Sidste år, de skabte en XANES-database for at løse den lokale struktur af en amorf titaniumoxidbelægning til fotokatalytiske applikationer. De byggede også en maskinlæringsmodel, der er i stand til at forudsige den lokale symmetri af absorberende atomer fra simulerede XANES-spektre af overgangsmetaloxider.
"Når du udfører spektral fortolkning baseret på domæneekspertise, vi har en tendens til at fokusere på specifikke funktioner, der er udviklet ud fra vores intuition, " sagde Lu. "Maskinlæring kan udtrække den information, vi har brug for på en statistisk fremtrædende måde, der eliminerer menneskelig skævhed."
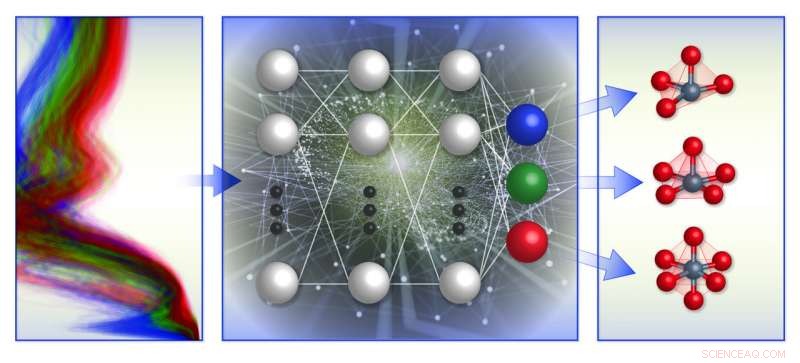
En skematisk illustration af holdets spektrumbaserede lokale kemiske miljøklassificeringsramme. De trænede maskinlæringsmodeller (midten) med computerbaseret røntgenabsorptionsspektradatabase (venstre) til at forudsige den lokale geometri omkring positivt ladede overgangsmetalioner (højre). Kredit:Brookhaven National Laboratory
Forudsigelse af røntgenabsorptionsspektre
Bygger på deres tidligere succeser, holdet påtog sig et mere udfordrende problem:træne en maskinlæringsmodel til hurtigt at forudsige spektre baseret på kendte molekylære strukturer. En sådan model ville omgå behovet for beregningsmæssigt dyre simuleringer, som ikke er gennemførlige under operando-eksperimenter, når forskere studerer materialer under driftsforhold. På trods af voksende maskinlæringsbestræbelser på at forudsige materialers kemiske egenskaber, direkte forudsigelser af de spektrale funktioner af virkelige materialer var endnu ikke opnået.
"En teknisk vanskelighed er at opbygge en optimal repræsentation af molekylære strukturer, der kan kode den iboende symmetri af molekylerne som inputfunktioner til maskinlæringsmodellen, " sagde Yoo.
Ved at vedtage en nylig idé foreslået af videnskabsmænd hos Google, Topsakal og Carbone byggede en maskinlæringsmodel baseret på en grafrepræsentation af molekyler som input, hvor atomer er repræsenteret som noder og kemiske bindinger som kanter.
"Computere kan ikke se molekyler, som vi gør, " sagde Topsakal. "En graf er en naturlig måde at kode strukturen og forbindelsen af et molekyle på - at fange hvilke atomer der er forbundet og typen og længden af de kemiske bindinger, der forbinder dem. I øvrigt, denne repræsentation er invariant i forhold til transformationer såsom translationer og rotationer. Dette koncept er analogt med det inden for billedgenkendelse, hvor et objekt som en kat eller hund i en baggrund stadig kan klassificeres korrekt efter billedet er transformeret."
For at træne modellen til en proof-of-princip demonstration, holdet brugte en veletableret database (kaldet QM9) indeholdende beregnede strukturelle og kemiske oplysninger om 134, 000 små molekyler med op til ni tunge atomer pr. atomtype (kulstof, nitrogen, ilt, og fluor). Fra denne database, de valgte to træningsundersæt - en undergruppe med molekyler indeholdende mindst et oxygenatom, og en anden undergruppe med molekyler indeholdende mindst et nitrogenatom - og beregnede deres tilsvarende XANES-spektre. Derefter, de brugte deres trænede modeller til at forudsige XANES-spektrene for ilt- og nitrogenabsorptionskanter svarende til excitationer af elektroner i den inderste skal af de respektive atomer.
Maskinlæringsmodellen gengav næsten alle de signifikante absorptionstoppe og forudsagde toppositionerne (energier, hvor peaks optræder) og højder (absorptionsintensiteter) med meget høj nøjagtighed. Modellen opfangede også automatisk domænets viden om, at røntgenabsorptionsspektroskopi er følsom over for funktionelle grupper, eller grupper af atomer med lignende kemiske egenskaber og reaktivitet. Afhængigt af hvilken funktionel gruppe absorbatoratomet tilhører, forskellige funktioner optræder i spektrene.
"Vi er de første til at demonstrere, at en maskinlæringsmodel kan bruges til nøjagtigt at forudsige fulde spektrale funktioner af rigtige fysiske systemer direkte fra deres strukturer, " sagde Topsakal. "Selvom vi fokuserede på røntgenabsorptionsspektroskopi i vores undersøgelse, denne metode kunne generaliseres til at forudsige spektral information for andre populære teknikker, inklusive infrarød og gammastrålespektroskopi."
"Når vi træner maskinlæringsmodellen, vi behøver ikke at køre tidskrævende fysiske simuleringer, som tager minutter, timer, eller endda dage, " sagde Yoo. "Vi aktiverede ikke kun forudsigelse af spektra i realtid, men også den samtidige generering af hundreder og tusinder af spektreslutninger ved at bruge flere grafikbehandlingsenheder, eller GPU'er. En sådan teknologi er nøglen til at muliggøre automatiseret strålelinjekontrol og accelerere videnskabelig opdagelse. Kombineret med metoder til at prøve materialestrukturer, sådanne modeller kan bruges til hurtigt at screene relevante strukturer for at drive materialedesign og opdagelse."
Næste, holdet vil gerne kombinere koncepter fra deres model, der forudsiger lokal symmetri fra XANES-spektre, og denne nye model, der forudsiger XANES-spektre fra molekylære strukturer. Ultimativt, deres mål er at udtrække mere omfattende information om det lokale kemiske miljø eller endda strukturen af hele molekyler fra eksperimentelle målinger.
"Maskinlæringsværktøjer, såsom dem til billed- og talegenkendelse og opdagelse af stoffer, er under rivende udvikling, " sagde Lu. "Nøglen er at finde ud af, hvordan man tilpasser disse værktøjer på en innovativ måde for at tackle materialevidenskabelige problemer."
"Vores mål med at udvikle kunstig intelligens og maskinlæringsteknologier er at løse unikke videnskabelige udfordringer ved både at adoptere de nyeste teknologiske gennembrud på disse områder og komme med nye tilgange, der bidrager tilbage til de respektive forskningsmiljøer, " tilføjede Yoo.
 Varme artikler
Varme artikler
-
 Video:Sådan får kimchi sit kickKredit:The American Chemical Society Kimchi, den fermenterede kålskål, der er elsket i Korea og rundt om i verden, har en signatur skarp, sur tang. Disse unikke smag kommer fra ikke kun salt og k
Video:Sådan får kimchi sit kickKredit:The American Chemical Society Kimchi, den fermenterede kålskål, der er elsket i Korea og rundt om i verden, har en signatur skarp, sur tang. Disse unikke smag kommer fra ikke kun salt og k -
 Fremstilling af syrechloridprækursorer ved hjælp af shuttle-katalyseTilgangen drager fordel af en isodesmisk shuttle-katalyseproces for at undgå behovet for CO og HCl og overvinde den termodynamiske udfordring med at fremstille meget reaktive produkter. Kredit: Naturk
Fremstilling af syrechloridprækursorer ved hjælp af shuttle-katalyseTilgangen drager fordel af en isodesmisk shuttle-katalyseproces for at undgå behovet for CO og HCl og overvinde den termodynamiske udfordring med at fremstille meget reaktive produkter. Kredit: Naturk -
 Blød og ionledende hydrogel kunstig tunge til astringens opfattelseÆndringer i porestørrelse på mennesketunge efterlignede hydrogel under TA-behandling. (A) SEM-billeder af human tunge efterlignet hydrogel før TA-behandling. (B) Histogram af de gennemsnitlige mikropo
Blød og ionledende hydrogel kunstig tunge til astringens opfattelseÆndringer i porestørrelse på mennesketunge efterlignede hydrogel under TA-behandling. (A) SEM-billeder af human tunge efterlignet hydrogel før TA-behandling. (B) Histogram af de gennemsnitlige mikropo -
 Ny metodologi hjælper med at studere lovende målrettet lægemiddelleveringsstilladsForskere undersøgte, hvor mutationer var tilladt i den fodbold-lignende struktur lavet af MS2-stilladset. Kredit:Northwestern University Northwestern Engineering-forskere har udviklet en ny måde a
Ny metodologi hjælper med at studere lovende målrettet lægemiddelleveringsstilladsForskere undersøgte, hvor mutationer var tilladt i den fodbold-lignende struktur lavet af MS2-stilladset. Kredit:Northwestern University Northwestern Engineering-forskere har udviklet en ny måde a
- Økologiske landbrugsfordele i biodiversitet og overskud afhænger af beliggenhed
- Tesla, andre hjælper puertoricanere med at gå på solskin i kraft af uro
- At bestå et udfordrende indledende kemikursus giver det største løft til underrepræsenterede stu…
- CERN:Undersøgelse kaster lys over et af fysikkens største mysterier - hvorfor der er mere stof end…
- Hvordan er vand en vedvarende ressource?
- Synkroniseret kunstbelysning af fluktuationstypen