


Atomopløsningsproteinmodeller afslører nye detaljer om proteinbinding
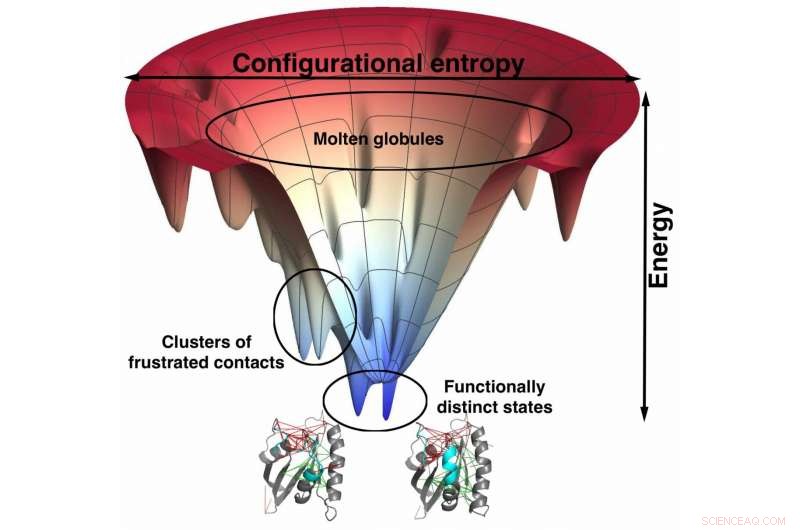
Atomskalamodeller af Rice University-forskere baseret på dem, der bruges til at forudsige, hvordan proteiner folder, viser en stærk sammenhæng mellem minimalt frustrerede bindingssteder og lægemiddelspecificitet. Tragten, en visuel repræsentation af proteinets energilandskab, når det foldes, hjælper med at finde de frustrerede websteder. Sådanne modeller kan føre til bedre designede lægemidler med færre bivirkninger. Kredit:Mingchen Chen/Rice University
At vide præcist, hvor proteiner er frustrerede, kan gå langt i retning af at lave bedre lægemidler.
Det er et resultat af en ny undersøgelse foretaget af Rice University-forskere, der leder efter de mekanismer, der stabiliserer eller destabiliserer nøglesektioner af biomolekyler.
Atomskalamodeller af risteoretikeren Peter Wolynes, hovedforfatter og alumnus Mingchen Chen og deres kolleger ved Center for Teoretisk Biologisk Fysik viser, at ikke blot er nogle specifikke frustrerede sekvenser i proteiner nødvendige for at tillade dem at fungere, at lokalisere dem giver også ledetråde til at opnå bedre specificitet for stoffer.
Den viden kan også hjælpe med at designe lægemidler med færre bivirkninger, Sagde Wolynes.
Teamets open-access undersøgelse vises i Naturkommunikation .
Atom-skala modellerne nul ind på interaktioner inden for mulige bindingssteder snarere end langt størstedelen af interaktionerne i proteiner, der styrer deres foldning. De finere opløsningsmodeller tillader inkorporering af co-faktorer som kemisk aktive ligander, herunder lægemiddelmolekyler. Forskerne siger, at denne evne giver ny indsigt i, hvorfor ligander bedst kun fanges af specifikke proteiner og ikke af andre.
"Unaturlige ligander, "alias stoffer, har tendens til at binde bedst med de frustrerede lommer i proteiner, der bliver minimalt frustrerede, når først stofferne binder, sagde Wolynes. At have en måde at finde og derefter lære detaljerne på disse minimalt frustrerede websteder ville hjælpe farmaceutiske virksomheder med at eliminere en masse forsøg og fejl.
"Standardmåden for at lave lægemiddeldesign er at prøve 10, 000 bindingssteder på et protein for at finde dem, der passer, " sagde Wolynes. "Vi siger, at du ikke behøver at prøve alle mulige bindingssteder, bare et rimeligt rimeligt tal for at forstå statistikken over, hvad der kunne fungere i lokale miljøer.
"Det er forskellen mellem at tage en meningsmåling og faktisk at have et valg, " sagde han. "Afstemningen er billigere, men du bliver stadig nødt til at tjekke tingene ud."
Rice-forskerne er kendt for deres energilandskabsteori om, hvordan proteiner folder. Det anvender normalt grovkornede modeller, hvor aminosyrer er repræsenteret af blot nogle få steder.
Den strategi kræver mindre computerkraft end at forsøge at bestemme positionerne over tid af hvert atom i hver rest, og alligevel har det vist sig meget præcist i at forudsige, hvordan proteiner folder baseret på deres sekvenser. Men for denne undersøgelse, forskerne modellerede proteiner og protein-ligandkomplekser på atomniveau for at se, om de kunne finde ud af, hvordan frustration giver nogle dele af et protein den fleksibilitet, der kræves for at binde sig til andre molekyler.
"En af de fantastiske ting ved modellering ved alle-atom opløsning er, at det giver os mulighed for at evaluere, om lægemiddelmolekyler passer godt ind i bindingssteder eller ej, " sagde Wolynes. "Denne metode er i stand til hurtigt at vise, om et bindingssted for et bestemt lægemiddel vil være minimalt frustreret eller vil forblive en frustreret region. Hvis stedet efter at molekylet binder forbliver frustreret, proteinet kunne omarrangere, eller lægemidlet kunne ændre sin orientering på en sådan måde, at det kunne give anledning til bivirkninger."
Ved at modellere de frustrerede websteder - og nogle gange ændre dem for at se, hvad der ville ske - lader forskerne se, hvordan lægemiddelspecificitet korrelerer med bindingslommer. Frustrationsanalyse, de skrev, giver "en rute til screening for mere specifikke forbindelser til lægemiddelopdagelse."
"Dette frustrationsbegreb var der helt i begyndelsen af vores arbejde med proteinfoldning, " sagde Wolynes. "Da vi anvendte det på rigtige proteinmolekyler, vi fandt nogle eksempler, hvor foldemekanismen overtrådte, hvad vi ville forudsige fra en perfekt tragt. Derefter opdagede vi, at disse afvigelser fra tragtebilledet opstod, hvor proteinet var, faktisk, lidt frustreret.
"Det var ligesom undtagelsen, der beviser reglen, " sagde han. "Noget, der hele tiden er sandt, kan være trivielt. Men hvis det ikke er sandt 1% af tiden, det er et problem, der skal løses, og vi har været i stand til at gøre det med AWSEM, vores software til strukturforudsigelse."
Det er muligt at udvide softwaren til at analysere frustration på atomniveau, som beskrevet af gruppen i et andet nyligt papir. Men de beregningsmæssige omkostninger ved at spore hvert atom i et protein er så høje, at forskerne havde brug for en måde at prøve bevægelserne i specifikke områder, hvor frustration kan forvirre foldningsruten.
"Mingchen indså, at der var en effektiv algoritme til at prøve de lokale miljøer på bindingssteder, men bevare den atomistiske opløsning, " sagde Wolynes, hvem bemærkede han og Chen, nu i den private industri, bruger modellerne til at undersøge mulige behandlingsmetoder, herunder COVID-19-relaterede lægemidler.
 Varme artikler
Varme artikler
-
 Bedre sammen:Sammensmeltet mikroskop giver et hidtil uset kig på biologiske processerDe hurtige bevægelser af Rab11-partikler kan tydeligt afbildes med det nye øjeblikkelige TIRF-SIM-mikroskop. Kredit:Hari Shroff, National Institute of Biomedical Imaging and Bioengineering Forsker
Bedre sammen:Sammensmeltet mikroskop giver et hidtil uset kig på biologiske processerDe hurtige bevægelser af Rab11-partikler kan tydeligt afbildes med det nye øjeblikkelige TIRF-SIM-mikroskop. Kredit:Hari Shroff, National Institute of Biomedical Imaging and Bioengineering Forsker -
 Når vi kan fange CO2 -emissioner, her er hvad vi kunne gøre med detDen foreslåede tidslinje for CO2 -udnyttelsesmetoder. Kredit:Bushuyev og De Luna et al./Joule De tusinder af tons kuldioxid (CO2), der udsendes fra kraftværker hvert år, behøver ikke at gå i atmos
Når vi kan fange CO2 -emissioner, her er hvad vi kunne gøre med detDen foreslåede tidslinje for CO2 -udnyttelsesmetoder. Kredit:Bushuyev og De Luna et al./Joule De tusinder af tons kuldioxid (CO2), der udsendes fra kraftværker hvert år, behøver ikke at gå i atmos -
 Vegetabilske proteiner erstatter petroleumsbaserede råvarerRapsmark i blomst:Raps er det mest dyrkede oliefrø i Tyskland – det er på tredjepladsen på verdensplan efter oliepalme og soja. Kredit:Alexas_Fotos/pixabay.com Ligesom cellulose, lignin og fedtsto
Vegetabilske proteiner erstatter petroleumsbaserede råvarerRapsmark i blomst:Raps er det mest dyrkede oliefrø i Tyskland – det er på tredjepladsen på verdensplan efter oliepalme og soja. Kredit:Alexas_Fotos/pixabay.com Ligesom cellulose, lignin og fedtsto -
 Ny forskning baner vejen for simulering af katalysatorer under reaktionsbetingelserEn illustration af nanopartikler under reaktionsbetingelser blev vist på forsiden af ACS Catalysis. Kredit:Raffaele Cheula Beregningsmæssig katalyse, et felt, der simulerer og accelererer opdage
Ny forskning baner vejen for simulering af katalysatorer under reaktionsbetingelserEn illustration af nanopartikler under reaktionsbetingelser blev vist på forsiden af ACS Catalysis. Kredit:Raffaele Cheula Beregningsmæssig katalyse, et felt, der simulerer og accelererer opdage
- Hvordan man driver elektronik ved hjælp af mekanisk bevægelse
- Gammel malariabehandling viste sig at forbedre leveringen af nanopartikler til tumorer
- Forbud mod slagvåben reducerer masseskydning markant
- Vi skal tale:Kommunikation forhindrer passivitet ved at udnytte goodwill
- Ny biofarmaceutisk kvalitetskontrolmetode i test
- Magnonic nanoantenner:optisk inspireret databehandling med spin-bølger et skridt nærmere