


Gentænke spinkemi fra et kvanteperspektiv
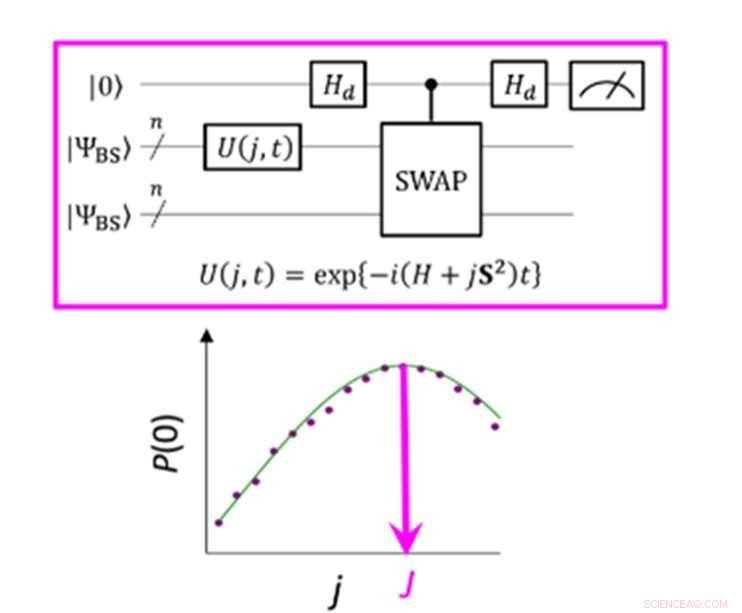
Et kvantekredsløb, der muliggør den maksimale sandsynlighed for P(0) ved måling af parameteren J. Kredit:K. Sugisaki, K. Sato og T. Takui
Forskere ved Osaka City University bruger kvantesuperpositionstilstande og Bayesiansk inferens til at skabe en kvantealgoritme, let eksekverbar på kvantecomputere, der nøjagtigt og direkte beregner energiforskelle mellem den elektroniske jord og exciterede spintilstande af molekylære systemer i polynomiel tid.
At forstå, hvordan den naturlige verden fungerer, gør os i stand til at efterligne den til gavn for menneskeheden. Tænk på, hvor meget vi er afhængige af batterier. Kernen er forståelsen af molekylære strukturer og elektronernes adfærd i dem. At beregne energiforskellene mellem et molekyles elektroniske jord og exciterede spin-tilstande hjælper os med at forstå, hvordan man bedre kan bruge det molekyle i en række forskellige kemikalier, biomedicinske og industrielle anvendelser. Vi har gjort store fremskridt inden for molekyler med lukkede skalsystemer, hvor elektroner er parret og stabile. Open-shell systemer, på den anden side, er mindre stabile og deres underliggende elektroniske adfærd er kompleks, og dermed sværere at forstå. De har uparrede elektroner i deres grundtilstand, som får deres energi til at variere på grund af elektronspins iboende natur, og gør målinger vanskelige, især da molekylerne øges i størrelse og kompleksitet. Selvom sådanne molekyler er rigelige i naturen, der mangler algoritmer, der kan håndtere denne kompleksitet. En forhindring har været at håndtere det, der kaldes den eksponentielle eksplosion af beregningstid. At bruge en konventionel computer til at beregne, hvordan de uparrede spins påvirker energien af et åbent skal-molekyle, ville tage hundreder af millioner af år, tid mennesker ikke har.
Kvantecomputere er under udvikling for at hjælpe med at reducere dette til det, der kaldes "polynomisk tid". Imidlertid, den proces, videnskabsmænd har brugt til at beregne energiforskellene for åbne skal-molekyler, har stort set været den samme for både konventionelle og kvantecomputere. Dette hæmmer den praktiske brug af kvanteberegning i kemiske og industrielle applikationer.
"Tilgange, der påberåber sig ægte kvantealgoritmer, hjælper os med at behandle open-shell-systemer meget mere effektivt end ved at bruge klassiske computere, " oplyser Kenji Sugisaki og Takeji Takui fra Osaka City University. Sammen med deres kolleger, de udviklede en kvantealgoritme, der kan eksekveres på kvantecomputere, der kan, for første gang, nøjagtigt beregne energiforskelle mellem den elektroniske jord og exciterede spin-tilstande af molekylære systemer med åben skal. Deres resultater blev offentliggjort i tidsskriftet Kemisk Videnskab den 24. december 2020.
Energiforskellen mellem molekylære spin-tilstande er karakteriseret ved værdien af udvekslingsinteraktionsparameteren J. Konventionelle kvantealgoritmer har været i stand til nøjagtigt at beregne energier for molekyler med lukket skal "men de har ikke været i stand til at håndtere systemer med en stærk multi-konfigurationsmæssig Karakter, " fastslår gruppen. Indtil nu, forskere har antaget, at for at opnå parameteren J skal man først beregne den samlede energi for hver spin-tilstand. I molekyler med åben skal er dette vanskeligt, fordi den samlede energi i hver spin-tilstand varierer meget, da molekylet ændrer sig i aktivitet og størrelse. Imidlertid, "energiforskellen i sig selv er ikke meget afhængig af systemets størrelse, " bemærker forskerholdet. Dette fik dem til at lave en algoritme med beregninger, der fokuserede på spinforskellen, ikke de enkelte spin-tilstande. At skabe en sådan algoritme krævede, at de gav slip på antagelser udviklet fra mange års brug af konventionelle computere og fokuserede på de unikke karakteristika ved kvanteberegning – nemlig "kvantesuperpositionstilstande."
"Superposition" lader algoritmer repræsentere to variable på én gang, hvilket så tillader videnskabsmænd at fokusere på forholdet mellem disse variabler uden at skulle bestemme deres individuelle tilstande først. Forskerholdet brugte noget, der kaldes en brudt-symmetri bølgefunktion som en superposition af bølgefunktioner med forskellige spintilstande og omskrev den til Hamiltons ligning for parameteren J. Ved at køre dette nye kvantekredsløb, holdet var i stand til at fokusere på afvigelser fra deres mål og ved at anvende Bayesiansk inferens, en maskinlæringsteknik, de indbragte disse afvigelser for at bestemme udvekslingsinteraktionsparameteren J. "Numeriske simuleringer baseret på denne metode blev udført for den kovalente dissociation af molekylært hydrogen (H) 2 ), tredobbeltbindingsdissociationen af molekylært nitrogen (N 2 ), og grundtilstandene for C, Åh, Si-atomer og NH, Åh + , CH 2 , NF og O 2 molekyler med en fejl på mindre end 1 kcal/mol, " tilføjer forskerholdet.
"Vi planlægger at installere vores Bayesian eXchange koblingsparameterberegner med Broken-symmetry wave functions (BxB) software på kortsigtede kvantecomputere udstyret med støjende (ingen kvantefejlkorrektion) mellemskala (flere hundreder af qubits) kvanteenheder (NISQ-enheder) ), at teste anvendeligheden til kvantekemiske beregninger af faktiske betydelige molekylære systemer."
 Varme artikler
Varme artikler
-
 Sætte fejl på menuen, sikkertKredit:Pixabay/CC0 Public Domain Tanken om at spise insekter er mavedrejning for mange, men ny forskning fra Edith Cowan University (ECU) kaster lys over allergifremkaldende proteiner, som kan udg
Sætte fejl på menuen, sikkertKredit:Pixabay/CC0 Public Domain Tanken om at spise insekter er mavedrejning for mange, men ny forskning fra Edith Cowan University (ECU) kaster lys over allergifremkaldende proteiner, som kan udg -
 Fremtiden for elektroniske enheder:Stærke og selvhelbredende iongelerIongelen udviser hurtig selvhelbredende evne ved stuetemperatur ved at omdanne hydrogenbindinger på den beskadigede overflade. Brudspændingen af iongelen efter heling 3 timer er sammenlignelig med d
Fremtiden for elektroniske enheder:Stærke og selvhelbredende iongelerIongelen udviser hurtig selvhelbredende evne ved stuetemperatur ved at omdanne hydrogenbindinger på den beskadigede overflade. Brudspændingen af iongelen efter heling 3 timer er sammenlignelig med d -
 Klar til nærbilledet - en bakteries elektrontransportvejRespiration i Actinomycetes og overordnet arkitektur af Mycobacterial respiratorisk maskine CIII2CIV2SOD2. A) Den respiratoriske elektronoverførselskæde i Actinomycetes (venstre) og de 5 større prokar
Klar til nærbilledet - en bakteries elektrontransportvejRespiration i Actinomycetes og overordnet arkitektur af Mycobacterial respiratorisk maskine CIII2CIV2SOD2. A) Den respiratoriske elektronoverførselskæde i Actinomycetes (venstre) og de 5 større prokar -
 Denne hydrogeltablet kan rense en liter flodvand på en timeBrug af hydrogel-tabletten til at rense vand kræver ingen energitilførsel og skaber ikke skadelige biprodukter. Kredit:University of Texas i Austin. Så meget som en tredjedel af verdens befolkning
Denne hydrogeltablet kan rense en liter flodvand på en timeBrug af hydrogel-tabletten til at rense vand kræver ingen energitilførsel og skaber ikke skadelige biprodukter. Kredit:University of Texas i Austin. Så meget som en tredjedel af verdens befolkning
- Software til at simulere kommercielle atomreaktorer
- Hvordan man forhindrer katodisk korrosion af metalelektroder i elektroorganisk syntese
- Hvilke dyr spiser normalt Hamster i det vilde?
- Forskere udvikler nye nanopartikler, der effektivt og selektivt dræber kræftceller
- Hvordan COVID-19 forstyrrer og transformerer sportens fremtid
- Forskere offentliggør vandbøflens genom