


Simple entropier for komplicerede molekyler
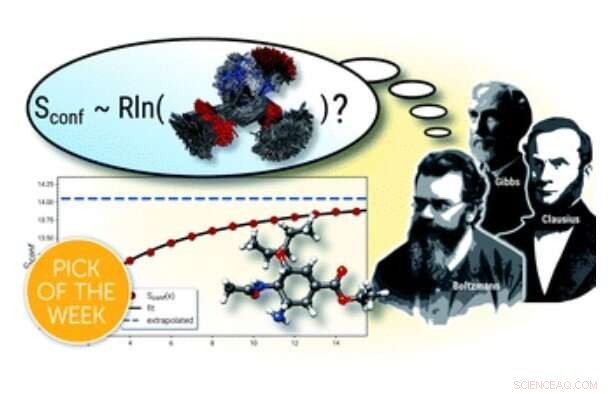
Grafisk abstrakt. Kredit:Beregning af absolutte molekylære entropier og varmekapaciteter gjort enkel, Kemisk Videnskab (2021). DOI:10.1039/D1SC00621E
Kemikere fra universitetet i Bonn udviklede et beregningsværktøj til analyse af konformationelle entropier af fleksible molekyler. Deres metode muliggør termodynamisk undersøgelse af komplicerede kemiske systemer ved kombination af moderne kvantekemiske og klassiske modeller. I et vellykket forsøg på forenklinger, vigtige bidrag til entropien kan beregnes med minimal brugerindblanding, selv på almindelige stationære computere. Resultaterne er publiceret i tidsskriftet Kemisk Videnskab og blev fremhævet som "Ugens udvalg"-artiklen.
Udtrykket "entropi" blev introduceret i 1865 af den tyske fysiker Rudolf Clausius, som senere arbejdede og var rektor ved universitetet i Bonn. 2022 vil være 200-året for hans fødselsdag, og videnskabelige begivenheder og fejringer er planlagt på universitetet i Bonn. Entropi er en af de mest fundamentale termodynamiske egenskaber ved stof og er almindeligvis forbundet med en tilstand af uorden eller usikkerhed. Over tid har konceptet også taget fat i statistisk mekanik, som banebrydende af berømte fysikere Josiah Gibbs og Ludwig Boltzmann, og i informationsteori. I dag, entropi er et aktivt forskningsområde på tværs af mange videnskabelige områder, herunder beregningskemi.
For molekyler bliver entropien vigtig som en del af den temperaturafhængige beskrivelse af det indre, såkaldt fri energi, hvoraf mange egenskaber såsom kemiske ligevægte eller reaktionshastigheder er afledt. I moderne beregningskemi, entropien af et molekyle opnås fra energiniveauer af atomare vibrationer i en molekylær struktur. Her, på grund af høje beregningsomkostninger på det kvantekemiske niveau adskillige teoretiske forenklinger, såsom den såkaldte stive-rotor harmoniske-oscillator tilnærmelse, skal indføres, og beregninger udføres for det meste kun for en enkelt struktur. For fleksible molekyler fører dette til forsømmelse af et vigtigt bidrag kaldet den konformationelle entropi, som beskriver den molekylære "lidelse" fra alle termisk tilgængelige konformationer. Sådanne fleksible tilfælde er almindelige og vigtige for mange farmaceutiske lægemidler.
I et nyligt forsøg på at give nøjagtige temodynamiske beskrivelser af fleksible molekyler, Prof. Dr. Stefan Grimme og kolleger fra Mulliken Center for Teoretisk Kemi ved Universitetet i Bonn udviklede et nyt beregningsværktøj til beregning af konformationelle entropier. Mens matematiske formuleringer til beregninger af den konformationelle entropi har været kendt i temmelig lang tid, et hovedproblem er at finde og evaluere det enorme antal mulige strukturer, der allerede når milliarder af mellemstore molekyler. Derfor, en kernekomponent i den nyligt introducerede og frit tilgængelige software er en effektiv algoritme til denne opgave, som fungerer med minimalt brugerinput, selv på almindelige stationære computere. For at opnå den nødvendige effektivitet, der blev anvendt semiempriske kvantekemiske metoder, som også er udviklet i Grimmes gruppe, sammen med standard kvantemekaniske beregninger. I artiklen blev det vist, at proceduren er i stand til at behandle selv store og ekstremt fleksible systemer med hidtil uset nøjagtighed for den molekylære entropi. Det er forfatterne, der håber, at den nye beregningsprotokol kan hjælpe med at opnå nøjagtige termodynamiske data mere rutinemæssigt, og at den vil finde udbredt anvendelse i beregningskemi.
Stefan Grimmes forskergruppe arbejder med aktuelle emner inden for kvantekemi med fokus på beregningseffektivitet og store molekyler. Hans kollega Philipp Pracht er i øjeblikket ved at færdiggøre sin ph.d. afhandling og er hovedforfatter af programmet CREST, der anvendes til de konformationelle entropiberegninger. Denne forskning er offentliggjort open access i Kemisk Videnskab , Royal Society of Chemistrys fagfællebedømte flagskibstidskrift.
 Varme artikler
Varme artikler
-
 Gør og ikke gør det i Science LabSelvom det kan være sjovt og spændende at lære ægte videnskab, kan farer også lurer i mange laboratoriesituationer. Vær meget opmærksom på den sikkerhedspraksis, der følger med dine laboratorieaktivit
Gør og ikke gør det i Science LabSelvom det kan være sjovt og spændende at lære ægte videnskab, kan farer også lurer i mange laboratoriesituationer. Vær meget opmærksom på den sikkerhedspraksis, der følger med dine laboratorieaktivit -
 En laserdrevet programmerbar berøringsfri transfertrykteknik(A) Skematisk illustration af den laserdrevne programmerbare berøringsfri transfertrykproces via et aktivt elastomerisk mikrostruktureret stempel. (B) Udskrivning af enkelt Si-blodplade og LED-chip på
En laserdrevet programmerbar berøringsfri transfertrykteknik(A) Skematisk illustration af den laserdrevne programmerbare berøringsfri transfertrykproces via et aktivt elastomerisk mikrostruktureret stempel. (B) Udskrivning af enkelt Si-blodplade og LED-chip på -
 Peptidhydrogeler kan hjælpe med at helbrede traumatiske hjerneskaderSelvsamlede peptid nanofibre, vist her, danne en hydrogel, der forbedrer overlevelsen af corticale neuroner efter en traumatisk hjerneskade hos rotter. Kredit:Biplab Sarkar og Vivek Kumar Trauma
Peptidhydrogeler kan hjælpe med at helbrede traumatiske hjerneskaderSelvsamlede peptid nanofibre, vist her, danne en hydrogel, der forbedrer overlevelsen af corticale neuroner efter en traumatisk hjerneskade hos rotter. Kredit:Biplab Sarkar og Vivek Kumar Trauma -
 Forbedrede vandafvisende overflader opdaget i naturenNye opdagelser om insekters nanostruktur, såsom øjet på en myg, kunne hjælpe med at udvikle forbedrede vandafvisende belægninger. Kredit:Ling Wang, Penn State Gennem undersøgelse af insektoverflad
Forbedrede vandafvisende overflader opdaget i naturenNye opdagelser om insekters nanostruktur, såsom øjet på en myg, kunne hjælpe med at udvikle forbedrede vandafvisende belægninger. Kredit:Ling Wang, Penn State Gennem undersøgelse af insektoverflad
- Talende tromme vist nøjagtigt at efterligne talemønstre i det vestafrikanske sprog
- Ofre-overlevere fra voldtægt føler ikke, at retfærdigheden er opfyldt, også selvom den anklagede…
- Grønt møder nano:Forskere skaber multifunktionelle nanorør ved hjælp af ikke-toksiske materialer
- Hvad gør kirkegårde skræmmende?
- Mus, fisk og fluer:dyrene sendes stadig ud i rummet
- Intel buzz handler om visning på power diet, øget batterilevetid