


Fra kemiske grafer til strukturer
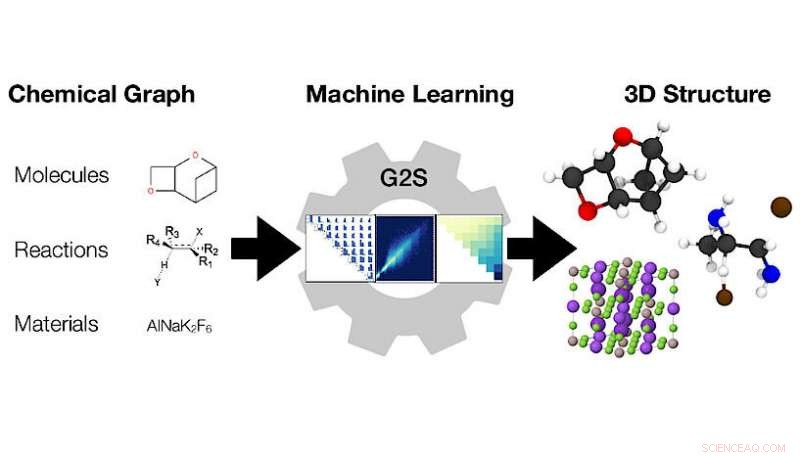
Maskinlæringsmodellen Graph2Structure bruger grafer af kemiske forbindelser (venstre) til at forudsige deres 3D-koordinater (højre). Kredit:Dominik Lemm, Universitetet i Wien
3D-konfigurationer af atomer dikterer alle materialers egenskaber. Kvantitative forudsigelser af nøjagtige ligevægtsstrukturer, 3D-koordinater for alle atomer, fra en kemisk graf, en repræsentation af strukturformlen, er en udfordrende og beregningsmæssigt dyr opgave, som er i begyndelsen af praktisk talt enhver computerkemi arbejdsgang. Forskere ved universitetet i Wien har nu udviklet en ny maskinlæringsbaseret model til at genveje dyre beregninger til direkte at forudsige strukturer ud fra grafer. Den nye metode til "Maskinlæring baseret energifri struktur forudsigelser af molekyler, overgangstilstande, og faste stoffer" præsenteres i det seneste nummer af Naturkommunikation .
Skønt almindeligvis afbildet som stiv, kemiske forbindelser er fleksible tredimensionelle objekter, der består af atomer, som konstant bevæger sig og oscillerer. Cyrus Levinthal bemærkede allerede i 1969, at den store mængde frihedsgrader for kemiske forbindelser formelt fører til et katastrofalt stort antal mulige konformationer godt op til 10, 300 (Levinthals paradoks). Inden for eksperimentelle observationer, imidlertid, 3D-konfigurationer af atomer svarer til veldefinerede frie energiminima og dikterer derved alle materialers egenskaber. Det paradigme, at struktur bestemmer funktion, er nøglen til at bestemme lægemiddelinteraktioner, optimering af katalysatorer eller reaktioner, og materialeopdagelse. Som en konsekvens, i de fleste computerbaserede screeningskampagner med høj kapacitet (en metode til hurtige videnskabelige eksperimenter), kun de mest stabile konfigurationer er eftertragtede. Afhængigt af niveauet af sofistikering inden for de tilnærmelser, der er foretaget ved estimering af materialers stabilitet, beregningsomkostninger kan variere fra minutter til timer eller endda dage for beregning af en enkelt struktur. I betragtning af det store rum med kemiske forbindelser, det rum, der er befolket af alle tænkelige forbindelser (estimeret til at overstige 1, 060) denne afvejning af omkostningskvalitet repræsenterer en stor flaskehals på området.
Forskere ved universitetet i Wien ledet af Anatole von Lilienfeld tacklede dette problem fra et andet perspektiv, udvikle en ny metode, der udnytter data og er universelt anvendelig til enhver form for kemi. Deres nye metode, Graph2Structure, bruger kvantekemiske data af høj kvalitet for at træne maskinlæringsmodeller, der er i stand til at forudsige nye 3D-strukturer til molekylære grafer af usete forbindelser. Denne direkte kortlægning af en molekylær graf til en specifik 3D-konfiguration gør det muligt for modellen effektivt at omgå enhver form for energiminimering, fører til en speedup på over en million sammenlignet med de konventionelle metoder. "Muligheden for at generere strukturer af høj kvalitet accelererer ikke kun molekylært design med høj gennemstrømning, men accelererer også den daglige arbejdsgang, " siger hovedforfatter af undersøgelsen i Naturkommunikation Dominik Lemm. "Plidelig generering af 3D-strukturer til selv eksotiske kemier, såsom open-shell-systemer eller overgangstilstande, er en af de sværeste opgaver inden for atomistisk simulering."
Yderligere resultater tyder på, at de genererede strukturer direkte kan bruges som input til efterfølgende evaluering af maskinlæringsbaserede egenskabsforudsigelsesmodeller, derved forbinder en molekylær graf til en strukturafhængig egenskab på en stringent og mere effektiv måde.
 Varme artikler
Varme artikler
-
 Sådan beregnes hydrogenionkoncentrationEn hydrogenionkoncentration i en opløsning er resultatet af tilsætning af en syre. Stærke syrer giver en højere koncentration af hydrogenioner end svage syrer, og det er muligt at beregne den resulter
Sådan beregnes hydrogenionkoncentrationEn hydrogenionkoncentration i en opløsning er resultatet af tilsætning af en syre. Stærke syrer giver en højere koncentration af hydrogenioner end svage syrer, og det er muligt at beregne den resulter -
 Sådan beregnes procentdelen af ionisering Givet pHpH-værdien af en opløsning er et mål for hydrogenionerne eller protonerne, der er til stede i den opløsning. Fordi syrer er protondonorer, hver dag, kan du sige, at en opløsning, der indeholder en s
Sådan beregnes procentdelen af ionisering Givet pHpH-værdien af en opløsning er et mål for hydrogenionerne eller protonerne, der er til stede i den opløsning. Fordi syrer er protondonorer, hver dag, kan du sige, at en opløsning, der indeholder en s -
 Kemi-gennembrud åbner nye døre for lægemiddeludviklere og kræftforskereKugle-og-stok model af cubaner. Kredit:CC0 To uafhængige kemigennembrud har åbnet et væld af døre, der tidligere var låst for lægemiddeludviklere og kræftforskere. Opdagelserne, som involverede a
Kemi-gennembrud åbner nye døre for lægemiddeludviklere og kræftforskereKugle-og-stok model af cubaner. Kredit:CC0 To uafhængige kemigennembrud har åbnet et væld af døre, der tidligere var låst for lægemiddeludviklere og kræftforskere. Opdagelserne, som involverede a -
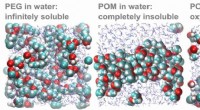 Opløselighedsmysteriet for udbredt plastik løstComputersimuleringer af PEG- og POM-molekyler i vand. Til venstre:PEG-molekyler opløses i vand. Center:POM-molekyler opløses ikke i vand, men hænge sammen i en klump. Til højre:Når ladningen af ilta
Opløselighedsmysteriet for udbredt plastik løstComputersimuleringer af PEG- og POM-molekyler i vand. Til venstre:PEG-molekyler opløses i vand. Center:POM-molekyler opløses ikke i vand, men hænge sammen i en klump. Til højre:Når ladningen af ilta
- Tiden tikker, når nationer mødes om klimaaftalen i Paris
- I Nigerias Lagos, akvatisk ukrudt plager vandveje
- Spansk taxistrejke mod apptjenester breder sig til Madrid
- NY vil bruge 6 millioner USD om året på computertræning for lærere
- Huawei:Amerikanske kontroller har ingen indflydelse, taler med Google
- Norwegian Air skærer 4, 700 ansatte i datterselskabskonkurs