


Neuralt netværk registrerer protein-peptidbindingssteder for at sætte gang i opdagelsen af peptidlægemidler
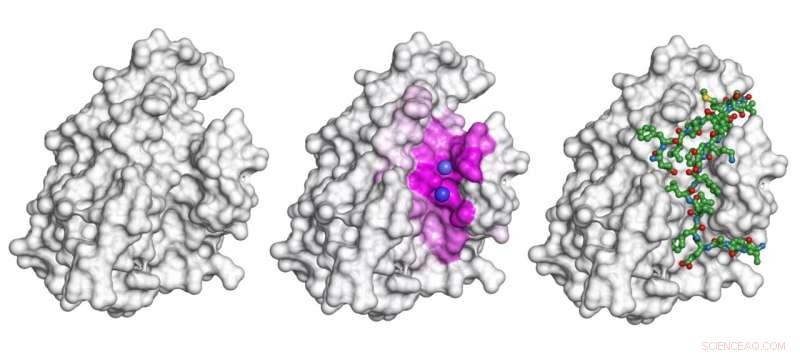
Den grå form er et protein. For scenariet med dette proteinbinding til peptidet vist som en grønlig stick-and-ball model til højre, modellen præsenteret i undersøgelsen fremhæver overfladen involveret i interaktionen (det lyserøde område i midten) og forudsiger de nøjagtige bindingssteder (lilla kugler). Kredit:Igor Kozlovskii og Petr Popov / Skoltech
To Skoltech-forskere har præsenteret en meget effektiv neural netværksmodel, der bruger data om strukturen af proteiner til at forudsige, hvilke af deres dele der interagerer med andre biologiske molekyler kaldet peptider. At vide, at dette er nyttigt til at udvikle lægemidler baseret på peptider, som kan påvirke protein-protein-interaktioner i celler på en målrettet og ikke-toksisk måde, regulerer en bred vifte af cellulære processer. Undersøgelsen udkom i Journal of Chemical Information and Modeling .
Proteiner er cellers maskineri, bevæger sig, engagere sig i hinanden, og kører alle mulige operationer. Farmakologer har altid været fascineret af udsigten til at pille ved interaktionerne mellem proteiner. Alligevel så de ud til at være off-limits som et potentielt lægemiddelmål:De større terapeutiske molekyler, kaldet biologiske lægemidler, kunne ikke trænge ind i cellen for at virke på proteiner, mens små molekylemidler ofte viste sig ude af stand til en sådan virkning.
Peptider, som naturligt medierer eller regulerer omkring 40 % af cellulære processer, indtager en lovende mellemvej og har udsigter til medicin rettet mod protein-protein-interaktioner. Peptider tilbyder det bedste fra begge verdener:Ligesom små molekyler, de kan trænge ind i cellemembranen for faktisk at nå deres mål, og de udviser også lav toksicitet, sammen med høj affinitet og specificitet (stærk og fokuseret handling) - kendetegnene for biologiske lægemidler.
At designe peptid-baserede lægemidler, farmakologer skal kende de såkaldte bindingssteder for et givet proteinmål. Det er, de pletter på proteinet, der kan binde sig til et peptid. Jo flere sådanne websteder er kendt, jo flere muligheder for lægemiddeldesign er tilgængelige.
Forskere kan identificere bindingssteder eksperimentelt, for eksempel, ved hjælp af røntgenkrystallografi, som afslører 3D-strukturen af krystalliserede proteiner ved at studere, hvordan de diffrakterer røntgenstråler. Men det er meget dyrt at gøre for en lang liste af molekyler, og beregningsmetoder tilbyder et hurtigere og billigere alternativ. Nogle af dem trækker på maskinlæringsteknikker, og efterhånden som flere data om strukturerne af protein-peptidkomplekser akkumuleres, disse metoder bliver mere kraftfulde og leverer stadig bedre forudsigelser om bindingssted.
I deres 22. juli papir i Journal of Chemical Information and Modeling , Skoltech Ph.D. studerende Igor Kozlovskii og adjunkt Petr Popov fra iMolecule-gruppen præsenterede en beregningsmetode kaldet BiteNetPp, som udnytter kraften i 3D-konvolutionelle neurale netværk til at detektere protein-peptidbindingssteder. I BiteNetPp, en kendt proteinstruktur tilføres et neuralt netværk, som derefter fremhæver formodede peptidbindingssteder, og udsender et sæt formodede 3D-koordinater, sammen med de tilhørende sandsynlighedsscore.
Petr Popov kommenterer tilgangen til bindingssteddetektion som billedgenkendelse, oprindeligt introduceret i holdets tidligere papir og overført til undersøgelsen rapporteret i denne historie:"Ligesom neurale netværk kan trænes til at genkende, sige, fodgængere eller cyklister på almindelige 2D-billeder, vi betragter bindingssteddetektion som at spotte en bestemt slags objekt i et billede. Forskellen er, at vi bruger 3D atomstrukturdata som vores input, så modellen fungerer på 'voxels, "en tredimensionel analog af pixels."
Den nyligt præsenterede model bygger faktisk på den i det foregående papir. "Dette kaldes domænetilpasning. BiteNetPp er den første model, der er blevet finjusteret på et protein-peptid-datasæt efter først at være blevet trænet på protein-små molekyle-data, " Popov forklarer. "Du kan forestille dig dette som at træne en model til at identificere steder, hvor cyklister har en tendens til at stoppe på gaden, men du begynder med data om, hvor fodgængere har en tendens til at stoppe - og først derefter udvider du dit domæne til cyklister. I stedet for at starte fra bunden, du omskoler modellen, forudse, at 'bindingsstederne' for cyklister kan dele nogle ligheder med dem, der tiltrækker fodgængere:du ved, isstandere, trafiklys, den slags."
Modellens skabere har vist, at BiteNetPp konsekvent udkonkurrerer eksisterende state-of-the-art metoder ved at sammenligne deres forudsigelser for de protein-peptidbindingssteder, der er kendt gennem eksperimentelle observationer. Vigtigt, den nye model tager mindre end et sekund at analysere en enkelt proteinstruktur, hvilket gør den velegnet til store undersøgelser. Der er tusindvis af protein-protein-interaktioner, der potentielt kan målrettes af peptid-baserede lægemidler, så beregningsmetoder skal være hurtige nok til at gøre deres screening mulig i en farmakologisk sammenhæng.
Sidste artikelTændt for IR-aktive organiske pigmenter
Næste artikelBayer taber endnu en appel af Roundup-kræftdommen
 Varme artikler
Varme artikler
-
 Nyt bevis indsendt til Grenfell Tower Inquiry om beklædningsreaktivitetKredit:University of Reading En kemiker fra University of Reading har skrevet til Grenfell Tower Inquiry efter kemiske tests på aluminiumsbeklædningspaneler, udført til et BBC-aktualitetsprogram.
Nyt bevis indsendt til Grenfell Tower Inquiry om beklædningsreaktivitetKredit:University of Reading En kemiker fra University of Reading har skrevet til Grenfell Tower Inquiry efter kemiske tests på aluminiumsbeklædningspaneler, udført til et BBC-aktualitetsprogram. -
 Når en defekt kan være gavnligVed hjælp af atomopløsningselektronmikroskopi, Arashdeep Singh Thind, en kandidatstuderende i Rohan Mishras laboratorium, studeret korngrænser i krystaller (se pile). Kredit:Washington University i St
Når en defekt kan være gavnligVed hjælp af atomopløsningselektronmikroskopi, Arashdeep Singh Thind, en kandidatstuderende i Rohan Mishras laboratorium, studeret korngrænser i krystaller (se pile). Kredit:Washington University i St -
 Ammoniumnitrat og jod:Et tilbageblik på den eksplosive historie med to essentielle stofferAmmoniumnitrat i granulær form er grundlaget for mange kvælstofgødninger. Kredit:Shutterstock Den frygtelige eksplosion, der ødelagde Beirut den 4. august, 2020, har trukket sig tilbage, men den f
Ammoniumnitrat og jod:Et tilbageblik på den eksplosive historie med to essentielle stofferAmmoniumnitrat i granulær form er grundlaget for mange kvælstofgødninger. Kredit:Shutterstock Den frygtelige eksplosion, der ødelagde Beirut den 4. august, 2020, har trukket sig tilbage, men den f -
 Justerbar emissiv organisk platformFigur viser designprincipperne for udvikling af en justerbar 2D COF, der er i stand til at udsende hvidt lys. Den frie intramolekylære bindingsrotation gør den grundlæggende molekylære enhed ikke-emit
Justerbar emissiv organisk platformFigur viser designprincipperne for udvikling af en justerbar 2D COF, der er i stand til at udsende hvidt lys. Den frie intramolekylære bindingsrotation gør den grundlæggende molekylære enhed ikke-emit
- Hvad frygter amerikanerne mest? Forskere udgiver 4. årlige Survey of American Fears
- Billede:Mystisk sydpolformation på Mars
- Nanokrystallinske formhukommelseslegeringer mister deres hukommelse, da de krystallinske korn bliver…
- Tilfældighed spiller en rolle i, hvordan sproget udvikler sig, undersøgelse finder
- Skjuler sig for et varmere klima i skoven
- Jordbaserede billeder af planeter taget af Pic-Net Pro-Am team