


Maskinlæring og kunstig intelligens hjælper med at forudsige kemiske reaktioners molekylære selektivitet
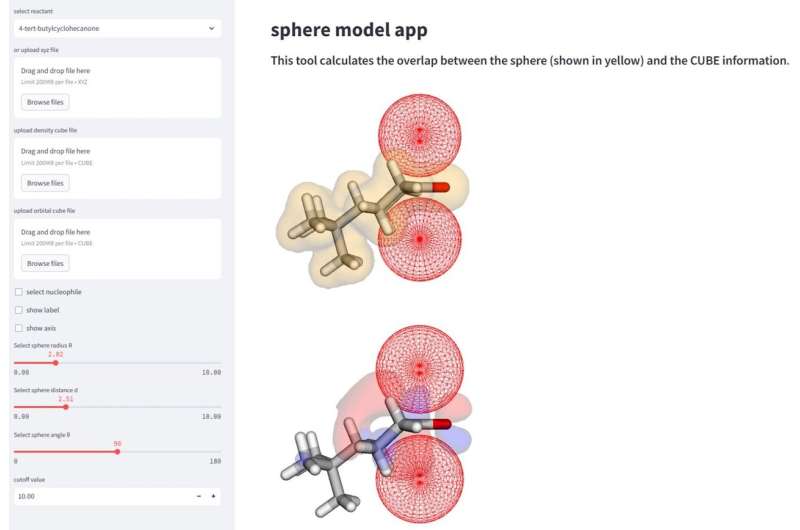
Der er få problemer nu, som AI og maskinlæring ikke kan hjælpe med at overvinde. Forskere fra Yokohama National University bruger denne moderne fordel til at løse, hvad konventionelle metoder ikke kan.
Der er mange regler at huske, når det kommer til vekselvirkningen mellem kulstofholdige (eller organiske) molekyler:placeringen af grupper på molekylet, der interagerer med dets miljø, størrelsen, formen og positionen af molekylet, og det molekyle, som det interagerer. Resultatet af en given reaktion kan være meget forskelligt afhængigt af disse faktorer og mange flere, og at forudsige disse udfald har vist sig at være noget af en udfordring på det kemiske område. Kontrol af resultatet er en meget nødvendig komponent i kemisk syntese, men forudsigelser er ikke altid nok.
Heldigvis kan maskinlæring og kunstig intelligens (AI) igen hjælpe med at skubbe fremskridt ved at forudsige hastigheden eller selektiviteten af en given reaktion. Derfor kan denne teknologi være nyttig til at forudsige, hvilket produkt der kan forventes.
Forskerne har offentliggjort deres resultater i Journal of Chemical Information and Modeling .
I organisk kemi er alle detaljer vigtige. To almindelige områder, der kan påvirke, hvordan et molekyle interagerer med andre molekyler, er steriske og orbitaler. Steriske refererer til arrangementet af molekyler, og steriske effekter kan bestemme formen og reaktiviteten af molekylet. Dette kan skyldes størrelsen eller ladningen af molekylet eller det enkelte atom. Orbitaler er en måde at forklare den mest sandsynlige placering af elektronerne, som igen kan interagere med andre molekyler eller atomer og forårsage reaktioner.
Disse faktorer kan ændre sig drastisk, hvor en nukleofil eller en elektrondonerende reaktant kan binde sig til modtagermolekylet. Dette er kendt som "selektivitet", og afhængigt af hvor molekylet hæfter, kan resultaterne danne forskellige produkter eller udbytter af det ønskede produkt. Forskere bruger kunstig intelligens og maskinlæring samt den nuværende viden om kemiske reaktioner til bedre at forklare disse aspekter af molekylær selektivitet.
"For at bestemme, hvilken information der kan bruges som væsentlig kemisk information, der skal gives til AI, er det nødvendigt at kombinere kemisk viden med viden om AI og maskinlæring," sagde den tilsvarende forfatter Hiroaki Gotoh, lektor ved det tekniske fakultet, Yokohama National University.
Først skulle computeren tilføres noget information, som man kunne lære fra. Information fra litteratur inden for beregningskemi og information fra tidligere undersøgelser blev brugt til at begynde undervisningsprocessen i AI. Efter en vis manuel dataindtastning for de specifikke molekyler, der blev brugt, og indstilling af optimale parametre, blev dataanalyser kørt baseret på de forudsagte resultater af testdatasættet. Disse analyser gør det muligt for AI at lære og forudsige fremtidige selektiviteter baseret på allerede kendt information.
"Denne metode muliggør en mere omfattende analyse og fortolkning af reaktionsmekanismer via beregning af parametrene for de sfæriske rum, der efterligner nærgående nukleofiler," sagde Daimon Sakaguchi, førsteforfatter af undersøgelsen ved afdelingen for kemi og biovidenskab, Yokohama National University.
Undersøgelsen forklarede med succes 323-reaktionsselektiviteten af otte nukleofiler, baseret på hvilket "ansigt" af molekylet, der ville give den mest ønskelige mængde produkt. Selektiviteten ændrer sig baseret på sterics af molekylet ud over dets orbitale faktorer. Forskere fandt ud af, at for nogle molekyler er orbitalfaktoren vigtigere til at bestemme ansigtets selektivitet, og andre er mere afhængige af molekylets steriske stoffer, når det interagerer med dets nukleofile.
Kombinationen af forudsigelig teknologi og maskinlæring med etableret viden om kemi kan give bedre resultater fra den kemiske reaktion og hjælpe kemikere med at syntetisere naturlige produkter og farmaceutiske kemikalier på en mere strømlinet måde.
Ved at strømline denne proces med brug af maskinlæring og kunstig intelligens kan der opstå flere eksperimenter. Ideelt set håber forskerne at samarbejde med eksperimentelle kemikere for at designe reaktioner, der vil fortsætte med udviklingen af mere forudsigelig teknologi til kemiske reaktioner.
Flere oplysninger: Daimon Sakaguchi et al., Brug af tredimensionel information til at forudsige og fortolke ansigtsselektiviteterne af nukleofile tilføjelser til cykliske ketoner, Journal of Chemical Information and Modeling (2024). DOI:10.1021/acs.jcim.4c00101
Journaloplysninger: Journal of Chemical Information and Modeling
Leveret af Yokohama National University
 Varme artikler
Varme artikler
-
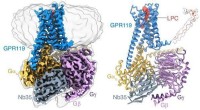 Undersøgelse afslører ligandgenkendelsesmekanisme for forældreløs receptor GPR119Identifikation af LPCet bundet til GPR119-Gs-komplekset ved hjælp af cryo-EM. Kredit:Peiyu Xu fra H. Eric Xus laboratorium Metaboliske sygdomme, herunder diabetes, fedtlever og fedme, er blevet en
Undersøgelse afslører ligandgenkendelsesmekanisme for forældreløs receptor GPR119Identifikation af LPCet bundet til GPR119-Gs-komplekset ved hjælp af cryo-EM. Kredit:Peiyu Xu fra H. Eric Xus laboratorium Metaboliske sygdomme, herunder diabetes, fedtlever og fedme, er blevet en -
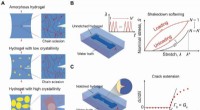 Anti-træthedsbrud hydrogelerDesignprincip for anti-træthedsbrud-hydrogeler. (A) Illustration af udmattelsesrevneudbredelse i en amorf hydrogel og i hydrogeler med lave og høje krystalliniteter under cykliske belastninger. De gul
Anti-træthedsbrud hydrogelerDesignprincip for anti-træthedsbrud-hydrogeler. (A) Illustration af udmattelsesrevneudbredelse i en amorf hydrogel og i hydrogeler med lave og høje krystalliniteter under cykliske belastninger. De gul -
 Hvordan kemiske reaktioner beregnesKredit:Pixabay/CC0 Public Domain Et enkelt molekyle indeholder et væld af information. Det inkluderer ikke kun antallet af hver type af atomer, men også hvordan de er arrangeret og hvordan de knyt
Hvordan kemiske reaktioner beregnesKredit:Pixabay/CC0 Public Domain Et enkelt molekyle indeholder et væld af information. Det inkluderer ikke kun antallet af hver type af atomer, men også hvordan de er arrangeret og hvordan de knyt -
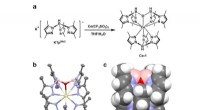 Dybblå organiske lysemitterende dioder baseret på et dublet-emission cerium(III) kompleksen. Syntetisk rute for komplekset. b. Enkeltkrystalstruktur af komplekset vist som ellipsoider ved 50 % sandsynlighedsniveau, hvor gul repræsenterer Ce, pink repræsenterer B, blå repræsenterer N, rød
Dybblå organiske lysemitterende dioder baseret på et dublet-emission cerium(III) kompleksen. Syntetisk rute for komplekset. b. Enkeltkrystalstruktur af komplekset vist som ellipsoider ved 50 % sandsynlighedsniveau, hvor gul repræsenterer Ce, pink repræsenterer B, blå repræsenterer N, rød
- Sådan måles gasledninger
- Computing med molekyler:Et stort skridt inden for molekylær spintronik
- Undersøgelse af våd brandrøg i skyer:Kan vand intensivere jordens opvarmning?
- Gamle tekster opmuntrede håb og udholdenhed, når de talte om endetiden
- Teoretisk fysiker forudsagde struktur af guldklynge, der hugger kuldioxid
- Historien om eksponenter