


Evolutionær algoritme genererer skræddersyede molekylære fingeraftryk
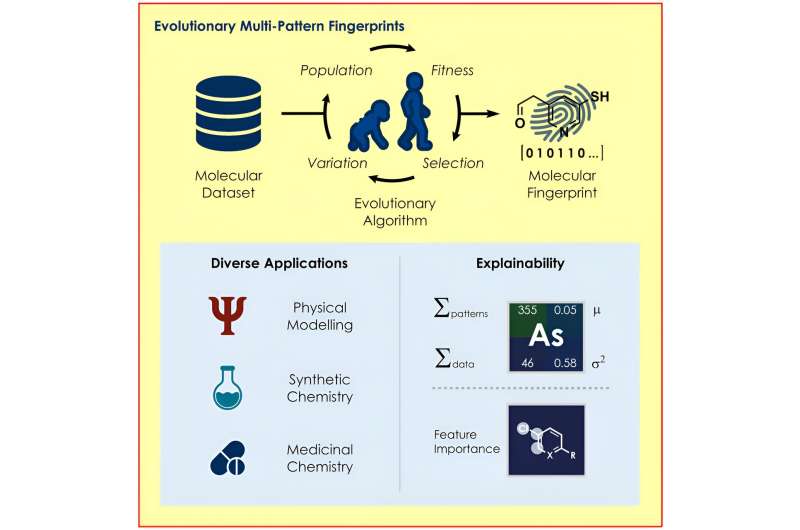
Et team ledet af prof Frank Glorius fra Institut for Organisk Kemi ved Universitetet i Münster har udviklet en evolutionær algoritme, der identificerer de strukturer i et molekyle, der er særligt relevante for et respektive spørgsmål, og bruger dem til at indkode molekylernes egenskaber for forskellige maskinlæringsmodeller.
Metoden er også velegnet til maskinel forudsigelse af kvantekemiske egenskaber og molekylers toksicitet. Det kan anvendes på ethvert molekylært datasæt og kræver ikke ekspertviden om de underliggende sammenhænge.
Kunstig intelligens og maskinlæring bliver mere og mere relevant i hverdagen – og det samme gælder kemi. Organiske kemikere er for eksempel interesserede i, hvordan maskinlæring kan hjælpe med at opdage og syntetisere nye molekyler, der er effektive mod sygdomme eller er nyttige på andre måder.
Den nye algoritme udviklet af Glorius' team søger efter optimale molekylære repræsentationer baseret på evolutionens principper ved hjælp af mekanismer som reproduktion, mutation og selektion. Afhængigt af modellen og det givne spørgsmål skabes der tilpassede "molekylære fingeraftryk", som kemikerne brugte i deres undersøgelse til at forudsige kemiske reaktioner med overraskende nøjagtighed.
Metoden, offentliggjort i tidsskriftet Chem , er også velegnet til at forudsige kvantekemiske egenskaber og molekylers toksicitet.
For at bruge maskinlæring skal forskerne først konvertere molekylerne til en computerlæsbar form. Mange forskergrupper har allerede taget fat på dette problem, og derfor er der forskellige måder at udføre denne opgave på. Det er dog svært at forudsige, hvilken af de tilgængelige metoder, der er bedst egnet til at besvare et specifikt spørgsmål – for eksempel at afgøre, om en kemisk forbindelse er skadelig for mennesker.
Den nye algoritme er designet til at hjælpe med at finde det optimale molekylære fingeraftryk i hvert enkelt tilfælde. For at gøre dette udvælger algoritmen gradvist de molekylære fingeraftryk, der opnår de bedste resultater i forudsigelsen fra mange tilfældigt genererede molekylære fingeraftryk.
"I overensstemmelse med naturens eksempel bruger vi mutationer, det vil sige tilfældige ændringer af individuelle komponenter i fingeraftrykkene, eller rekombinerer komponenter af to fingeraftryk," forklarer ph.d.-studerende Felix Katzenburg.
"I andre undersøgelser er molekyler ofte beskrevet af kvantificerbare egenskaber, som er blevet udvalgt og beregnet af mennesker," tilføjer Glorius.
"Da den algoritme, vi udviklede, automatisk identificerer de relevante molekylære strukturer, er der ingen systematiske skævheder forårsaget af menneskelige eksperter."
En anden fordel er, at metoden til kodning gør det muligt at forstå, hvorfor en model laver en bestemt forudsigelse. For eksempel er det muligt at drage konklusioner om, hvilke dele af et molekyle, der positivt eller negativt påvirker forudsigelsen af, hvordan en reaktion vil udspille sig, hvilket giver forskere mulighed for at ændre de relevante strukturer på en målrettet måde.
Münster-teamet fandt ud af, at deres nye metode ikke altid gav de mest optimale resultater.
"Når der er brugt betydelig menneskelig ekspertise på at udvælge særligt relevante molekylære egenskaber eller meget store mængder data er tilgængelige, har andre metoder såsom neurale netværk nogle gange fordelen," siger Katzenburg.
Et af undersøgelsens primære mål var dog at udvikle en metode til at indkode molekyler, som kan anvendes på ethvert molekylært datasæt og ikke kræver ekspertviden om de underliggende sammenhænge.
Flere oplysninger: Philipp M. Pflüger et al., En evolutionær algoritme til fortolkbare molekylære repræsentationer, Chem (2024). DOI:10.1016/j.chempr.2024.02.004
Journaloplysninger: Kem
Leveret af University of Münster
 Varme artikler
Varme artikler
-
 Forskere har syntetiseret ny flydende-krystallinsk fotokromLCD-tekstur observeret i et polarisationsoptisk mikroskop. Kredit:Alexey Bobrovsky Kemikere ved Lomonosov Moscow State University, i samarbejde med tjekkiske partnere, har syntetiseret og studeret
Forskere har syntetiseret ny flydende-krystallinsk fotokromLCD-tekstur observeret i et polarisationsoptisk mikroskop. Kredit:Alexey Bobrovsky Kemikere ved Lomonosov Moscow State University, i samarbejde med tjekkiske partnere, har syntetiseret og studeret -
 Forskere syntetiserer helbredende forbindelser i skorpiongiftStanford Kemi Professor Richard Zare holder den mexicanske skorpion art Diplocentrus melici i hans hånd. Kredit:Edson N. Carcamo-Noriega En skorpion hjemmehørende i det østlige Mexico kan have m
Forskere syntetiserer helbredende forbindelser i skorpiongiftStanford Kemi Professor Richard Zare holder den mexicanske skorpion art Diplocentrus melici i hans hånd. Kredit:Edson N. Carcamo-Noriega En skorpion hjemmehørende i det østlige Mexico kan have m -
 En platform til at forberede fluorescensmærkede proteiner og simulere deres native miljøVed at bruge en vaskemiddelfri metode, biologer kan fremstille fluorescens-mærkede proteiner sammen med et lille segment af den associerede cellemembran, bevare proteinets native miljø. Kredit:Jean-Ma
En platform til at forberede fluorescensmærkede proteiner og simulere deres native miljøVed at bruge en vaskemiddelfri metode, biologer kan fremstille fluorescens-mærkede proteiner sammen med et lille segment af den associerede cellemembran, bevare proteinets native miljø. Kredit:Jean-Ma -
 Forbedrede vandafvisende overflader opdaget i naturenNye opdagelser om insekters nanostruktur, såsom øjet på en myg, kunne hjælpe med at udvikle forbedrede vandafvisende belægninger. Kredit:Ling Wang, Penn State Gennem undersøgelse af insektoverflad
Forbedrede vandafvisende overflader opdaget i naturenNye opdagelser om insekters nanostruktur, såsom øjet på en myg, kunne hjælpe med at udvikle forbedrede vandafvisende belægninger. Kredit:Ling Wang, Penn State Gennem undersøgelse af insektoverflad
- Amerikanske prognosemænd:Her kommer endnu en varmere sommer end normal
- Nye simuleringer tyder på, at Jupiters fjerdestørste måne skyder vand ud fra sit underjordiske ha…
- Hvordan regulerer PA-, ABA- og CBF-veje synergistisk melonens kuldetolerance?
- Ny sanseteknologi kan hjælpe med at opdage sygdomme, svigagtig kunst, kemiske våben
- Sådan fungerer sorte huller
- Smog kvæler kulafhængigt Polen uden ende