


Katalysatorsøgning viser, hvordan computing kan fjerne gætteriet ud af kemi
Forestil dig at syntetisere og derefter teste over 50 forskellige komplekse molekyler for at identificere den mest effektive katalysator til en bestemt kemisk reaktion. Den traditionelle tilgang til at udvikle nye katalysatorer til kemiske reaktioner på denne "prøv det og se"-måde er ofte ekstremt arbejdskrævende, hvilket kræver adskillige gentagne eksperimenter med potentielle kandidatmolekyler. Den nu allestedsnærværende teknik med maskinlæring kan gøre denne opgave meget mere effektiv ved at forudsige katalysatorers ydeevne på forhånd baseret på teoretiske karakteristika.
I en undersøgelse offentliggjort i Nature Communications , brugte forskere fra Osaka University et computerbibliotek af molekyler, der er blevet syntetiseret sammen med molekyler, der er helt teoretiske i øjeblikket til at finde den bedste katalysator til en specifik kemisk reaktion.
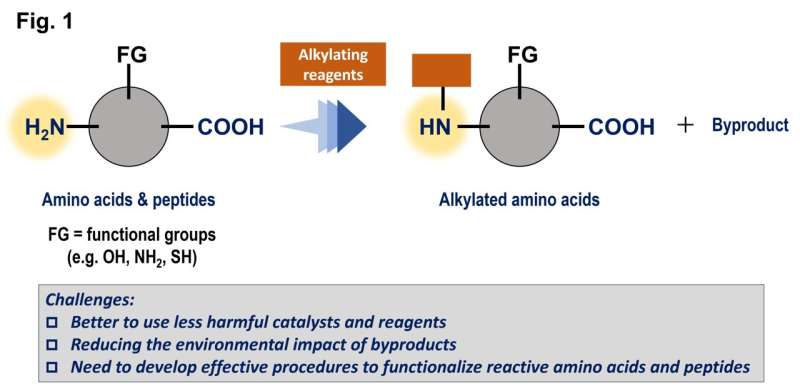
Formålet med arbejdet var at finde bedre måder at tilføje grupper af kulstof til aminosyrer og peptider, som er meget almindelige i levende organismer, for at modificere disse forbindelsers egenskaber. Som mange reaktioner forstærkes disse processer af katalysatorer, men en traditionel metalbaseret katalysator er ofte giftig og/eller dyr.
Denne undersøgelse havde til formål at bruge triarylboraner som katalysatorer, men på grund af deres relativt komplekse strukturer er der potentielt hundredvis af muligheder. Disse forbindelser er baseret på bor, som er et hovedgruppeelement, der er relativt billigt og mindre giftigt.
"Vurderingen af molekylære katalysatorer til organisk syntese kan være ekstremt tidskrævende," siger hovedforfatter af undersøgelsen Yusei Hisata. "I tilfældet med de triarylboraner, der bruges i vores arbejde, kan mange permutationer af molekylære strukturer kræve måneders undersøgelse blot for at identificere den optimale kandidat."
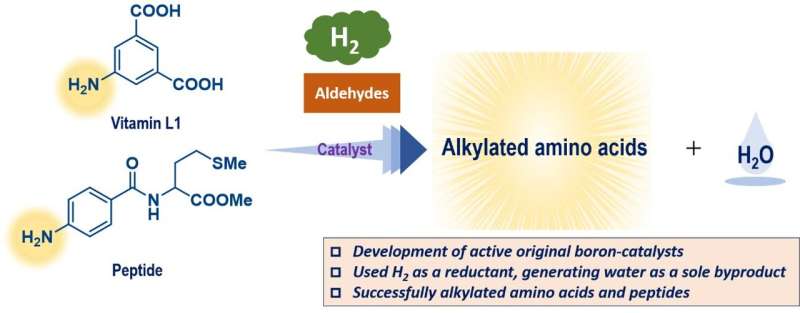
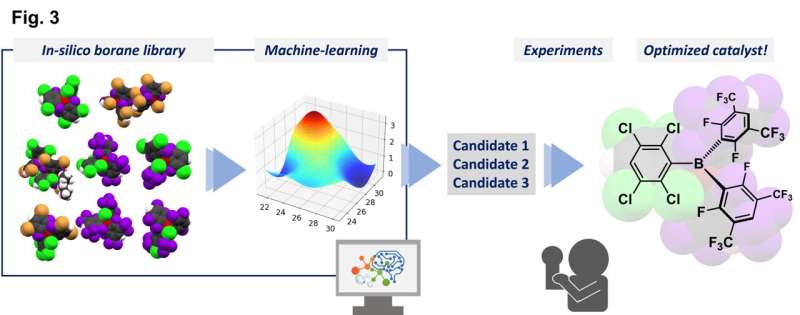
Forskerne kombinerede eksperimentelle data fra et begrænset antal syntetiserede triarylboraner med egenskaber forudsagt for andre molekyler, der endnu ikke er blevet syntetiseret, ved hjælp af teoretiske beregninger, for at lave et bibliotek med 54 mulige katalysatorer.
"Denne proces vurderede parametre, som vi forudsagde ville påvirke reaktionens fremskridt," forklarer Yoichi Hoshimoto, den tilsvarende forfatter. "Disse inkluderede faktorer såsom de molekylære orbitale energiniveauer og energibarriererne for visse processer."
En Gaussisk procesregression ved brug af in silico-biblioteket identificerede en lovende kandidat, og test med denne triarylboran viste et højt ydeevneniveau. Denne forbindelse kunne fremme reaktionerne af en aminosyre i meget høje udbytter og tolerere tilstedeværelsen af adskillige forskellige funktionelle grupper. Som en ekstra fordel genererer disse reaktioner kun vand som et harmløst biprodukt, fordi de med succes brugte molekylært hydrogen, H2 , som et reagens.
Dette arbejde undersøgte også andre måder at sænke processens miljøpåvirkning og fandt ud af, at det farlige opløsningsmiddel tetrahydrofuran kunne erstattes med det mindre giftige alternativ 4-methyltetrahydropyran.
Moderne kemikere står over for stigende krav, og de jonglerer med at udvikle nye synteser med begrænsede peers, mens de overvejer miljøpåvirkning, effektivitet, omkostninger, bæredygtighed og andre faktorer. Denne undersøgelse demonstrerer et vigtigt skridt fremad i brugen af maskinlæring til at strømline udviklingen af nye kemiske processer og fremhæver, hvordan disse nye processer kan inkorporere ændringer, der arbejder sammen om at generere grønne systemer.
Flere oplysninger: Yusei Hisata et al., In-silico-assisteret derivatisering af triarylboraner til katalytisk reduktiv funktionalisering af anilin-afledte aminosyrer og peptider med H2, Nature Communications (2024). DOI:10.1038/s41467-024-47984-0
Journaloplysninger: Nature Communications
Leveret af Osaka University
 Varme artikler
Varme artikler
-
 Mekanismen bag de elektriske ladninger genereret af fotosynteseFigur 1. Den båndlignende struktur af fotosystem II-komplekset (PDB ID:3ARC). Kredit:Kobe University Fotosyntese kræver en mekanisme til at producere store mængder kemisk energi uden at miste den
Mekanismen bag de elektriske ladninger genereret af fotosynteseFigur 1. Den båndlignende struktur af fotosystem II-komplekset (PDB ID:3ARC). Kredit:Kobe University Fotosyntese kræver en mekanisme til at producere store mængder kemisk energi uden at miste den -
 Designer peptider viser potentiale til at blokere vira, tilskynde til fremtidige studierKredit:Rensselaer Polytekniske Institut Kemisk fremstillede peptider, designet og udviklet af et team af forskere ved Rensselaer Polytechnic Institute, kunne vise sig at være værdifuld i kampen mo
Designer peptider viser potentiale til at blokere vira, tilskynde til fremtidige studierKredit:Rensselaer Polytekniske Institut Kemisk fremstillede peptider, designet og udviklet af et team af forskere ved Rensselaer Polytechnic Institute, kunne vise sig at være værdifuld i kampen mo -
 Chokoladefingeraftryk kunne bekræfte påstande på etikettenKredit:CC0 Public Domain Smagen og aromaen af en fin chokolade kommer frem fra dens økologi, ud over dens behandling. Men kan du være sikker på, at den bar, du har købt, virkelig er fra det ekso
Chokoladefingeraftryk kunne bekræfte påstande på etikettenKredit:CC0 Public Domain Smagen og aromaen af en fin chokolade kommer frem fra dens økologi, ud over dens behandling. Men kan du være sikker på, at den bar, du har købt, virkelig er fra det ekso -
 Forskere udvikler en metode til at undersøge millioner af potentielle egenproducerede lægemiddelka…Med en stor samling af fiskekroge, ETH-kemikere forsøger at fange fisken på en meget specifik måde, dvs. et molekylært mål. Kredit:ETH Zürich / Morris Köchle At lede efter nye stoffer er som at fi
Forskere udvikler en metode til at undersøge millioner af potentielle egenproducerede lægemiddelka…Med en stor samling af fiskekroge, ETH-kemikere forsøger at fange fisken på en meget specifik måde, dvs. et molekylært mål. Kredit:ETH Zürich / Morris Köchle At lede efter nye stoffer er som at fi
- Videospildesigner etablerer laboratorium for vedvarende energi
- Forskere laver smartphone-system til at teste for bly i vand
- Eksempler på diffusion i organer
- Hvordan lytning til tilfældig lyd kan låse op for et fanget sind
- Halloween er en vigtig måde at tænke døden på
- Sådan beregnes lift til rotorbladene