


Ny metode giver automatiseret beregning af overfladeegenskaber i krystaller
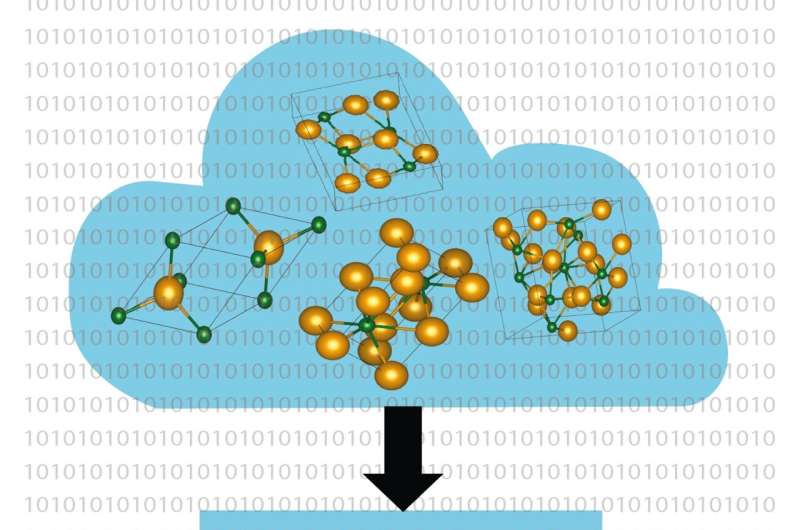
Computerbaserede metoder bliver et stadig mere kraftfuldt værktøj i jagten på nye materialer til nøgleteknologier som solceller, batterier og datatransmission. Prof. Dr. Caterina Cocchi og Holger-Dietrich Saßnick fra Universitetet i Oldenburg i Tyskland har nu udviklet en højkapacitetsautomatiseret metode til at beregne overfladeegenskaberne af krystallinske materialer, der starter direkte på niveauet af etablerede fysiklove (første principper).
I en artikel publiceret i tidsskriftet npj Computational Materials , rapporterer de, at dette kan fremskynde søgningen efter relevante materialer til applikationer på nøgleområder som energisektoren. De planlægger også at kombinere metoden med kunstig intelligens og maskinlæringsteknikker for at accelerere processen yderligere.
Hidtil har lignende metoder fokuseret på bulkmaterialer frem for overflader, forklarer de to fysikere. "Alle de relevante processer til energiomdannelse, produktion og lagring foregår på overflader," siger Cocchi, der leder forskningsgruppen Teoretical Solid State Physics ved University of Oldenburg.
At beregne overfladens materialeegenskaber er dog langt mere udfordrende end for komplette krystaller, fordi overfladefacetterne ofte har en kompleks struktur på grund af faktorer som defekter i krystalstrukturen eller ujævn vækst af en krystal, forklarer hun.
Denne kompleksitet skaber problemer for forskere inden for materialevidenskab:"Det er ofte ikke muligt klart at bestemme egenskaberne af prøver i eksperimenter," siger Cocchi. Dette motiverede Cocchi og hendes kollega Saßnick til at udvikle en automatiseret procedure til højkvalitetsscreening af nye forbindelsers egenskaber.
Plidelige resultater
Resultatet af deres arbejde blev indarbejdet i computerprogrammet aim2dat, som kun kræver den kemiske sammensætning af en forbindelse som input. Oplysningerne om krystallens struktur udvindes fra eksisterende databaser. Softwaren beregner derefter de forhold, hvorunder overfladen af materialet er kemisk stabil.
I det andet trin bestemmer den nøgleegenskaber, især den energi, der kræves for at excitere elektroner til ledningstilstande eller løsne sig fra en overflade. Denne parameter spiller en vigtig rolle i materialer, der f.eks. omdanner solenergi til elektricitet. "Vi laver ingen antagelser i vores beregninger; vi bruger kun kvantemekanikkens fundamentale ligninger, hvilket er grunden til, at vores resultater er meget pålidelige," forklarer Cocchi.
De to videnskabsmænd demonstrerede anvendeligheden af metoden ved hjælp af halvlederen cæsiumtellurid. Krystallerne af dette materiale, som bruges som elektronkilde i partikelacceleratorer, kan forekomme i fire forskellige former. "Sammensætningen og kvaliteten af materialeprøverne er svære at kontrollere i eksperimenter," bemærker Saßnick. Ikke desto mindre var Oldenburg-forskerne i stand til at udføre en detaljeret analyse af de fysiske egenskaber af de forskellige konfigurationer af cæsiumtellurid-krystallerne.
Cocchi og Saßnick har indlejret softwaren i et offentligt tilgængeligt programbibliotek, så andre forskere også kan bruge og forbedre proceduren. "Vores metode har et stort potentiale som et værktøj til at opdage nye materialer - og især fysisk og strukturelt komplekse faste stoffer - til alle slags anvendelser i energisektoren," siger Cocchi.
Flere oplysninger: Holger-Dietrich Saßnick et al., Automatiseret analyse af overfladefacetter:eksemplet med cæsiumtellurid, npj Computational Materials (2024). DOI:10.1038/s41524-024-01224-7
Leveret af University of Oldenburg
 Varme artikler
Varme artikler
-
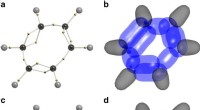 Efter 90 år, forskere afslører strukturen af benzenDVMS -strukturer til benzen. et Voronoi-sted til RHF/6-31G (d) bølgefunktion. Elektronpositionerne for et vilkårligt spin vises som små gule kugler. b Tværsnit gennem bølgefunktionen omkring Voronoi -
Efter 90 år, forskere afslører strukturen af benzenDVMS -strukturer til benzen. et Voronoi-sted til RHF/6-31G (d) bølgefunktion. Elektronpositionerne for et vilkårligt spin vises som små gule kugler. b Tværsnit gennem bølgefunktionen omkring Voronoi - -
 Sydafrika er et skridt tættere på forarbejdede titanlegeringerLavprislegeringer ville bane vejen for overkommelige medicinske implantater og proteser. Kredit:Monstar Studio/Shutterstock William Gregor, en amatør mineralog og kemiker, første gang opdagede ilm
Sydafrika er et skridt tættere på forarbejdede titanlegeringerLavprislegeringer ville bane vejen for overkommelige medicinske implantater og proteser. Kredit:Monstar Studio/Shutterstock William Gregor, en amatør mineralog og kemiker, første gang opdagede ilm -
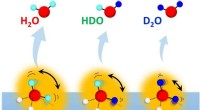 Indvirkningen af molekylær rotation på en ejendommelig isotopeffekt på vandhydrogenbindingerDesorption af vandisotopomerer (H 2 Åh, HDO og D 2 O) fra overflader af isotopblandet is med forskellige H/D-sammensætninger. Kredit:NINS/IMS Kvantenaturen af brintbindinger i vand manifeste
Indvirkningen af molekylær rotation på en ejendommelig isotopeffekt på vandhydrogenbindingerDesorption af vandisotopomerer (H 2 Åh, HDO og D 2 O) fra overflader af isotopblandet is med forskellige H/D-sammensætninger. Kredit:NINS/IMS Kvantenaturen af brintbindinger i vand manifeste -
 Forskere opdager ny transportrute for flygtige planteforbindelserEn petuniaknop, der ikke har åbnet sig (øverst), vil fra luften overføre flygtige forbindelser fra blomstens rør til stigmaet før åbning. Opdagelsen, af Purdue University videnskabsmænd, er den første
Forskere opdager ny transportrute for flygtige planteforbindelserEn petuniaknop, der ikke har åbnet sig (øverst), vil fra luften overføre flygtige forbindelser fra blomstens rør til stigmaet før åbning. Opdagelsen, af Purdue University videnskabsmænd, er den første
- Hvordan kunstig intelligens tager imod ransomware
- Undersøgelse samler den første observation af leptonisk henfald D+→ τ+ντ
- Vil der nogensinde være en lykkepille?
- Sådan konstrueres en 70 graders vinkel
- Forskere finder, at røg ved brande er mere afkøling på klimaet, end computermodeller antager
- Ældste opdagelse til dato af fysogastriske insekter