


Forskere afslører ny metode til at beregne mekaniske egenskaber af faste stoffer ved hjælp af maskinlæring
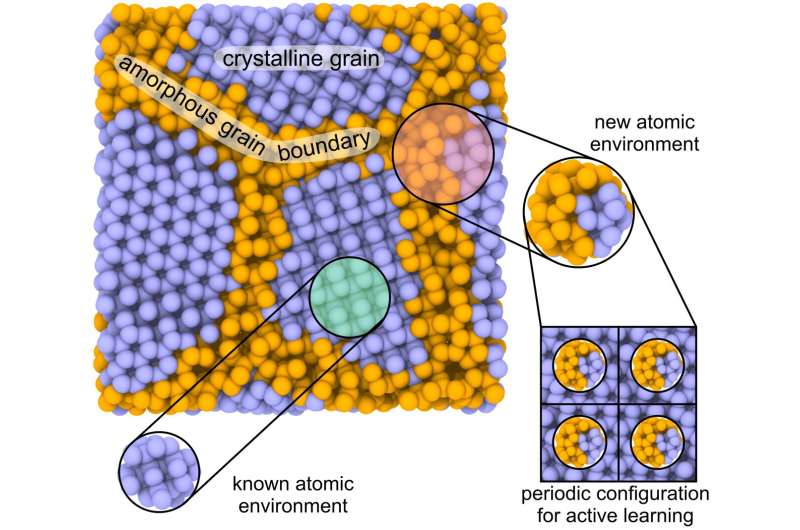
Et forskerhold fra Skoltech introducerede en ny metode, der udnytter maskinlæring til at studere egenskaberne af polykrystaller, kompositter og flerfasesystemer. Det opnåede høj nøjagtighed, næsten lige så god som kvantemekaniske metoder, som kun er anvendelige til materialer med mindre end et par hundrede atomer.
Den nye metode nyder også godt af aktiv læring på lokale atomare miljøer. Artiklen er udgivet i Advanced Theory and Simulations journal.
"Mange industrielle materialer syntetiseres som polykrystaller eller flerfasesystemer. De indeholder både en enkelt krystal og amorfe komponenter mellem enkeltkrystalkorn. Det store antal atomer gør det svært at beregne egenskaberne af disse systemer ved hjælp af moderne kvantemekaniske metoder. Tæthedsfunktionel teori kan kun anvendes på materialer med nogle få hundrede atomer."
"For at løse problemet bruger vi en maskinlæringstilgang baseret på Moment Tensor Potentials (MTP). Disse potentialer er også blevet udviklet på Skoltech under vejledning af professor Alexander Shapeev," kommenterede Faridun Jalolov, den førende forfatter af undersøgelsen og en Skoltech Ph.D. studerende på Materials Science and Engineering-uddannelsen.
Sammenlignet med andre løsninger ser forfatterne potentialet i den nye metode i aktiv læring på lokale atomare miljøer. Når man beregner en stor struktur med mange hundrede tusinde atomer, identificerer MTP, hvilket atom der laver en fejl i beregningerne, eller er beregnet forkert. Årsagen til dette kan være det begrænsede træningsdatasæt, som forhindrer alle mulige systemkonfigurationer i at blive overvejet.
Et lokalt miljø af dette atom bliver derefter "skåret ud", og dets energi beregnes ved hjælp af kvantemekanik. Bagefter føjes dataene tilbage til træningssættet for yderligere læring. Efterhånden som on-the-fly-læringen skrider frem, fortsætter beregningerne, indtil de støder på en anden konfiguration, der skal indgå i træningsprocessen. Andre kendte maskinlæringspotentialer kan ikke læres på små lokale dele af store strukturer, hvilket begrænser deres anvendelighed og nøjagtighed.
"Som et eksempel har vi undersøgt de mekaniske egenskaber af diamantpolykrystaller, som er de hårdeste naturligt forekommende materialer og ofte bruges i industrien - for eksempel ved fremstilling af boreudstyr til oliebrønde. Resultaterne viser, at de mekaniske egenskaber af disse polykrystallinske diamanter afhænger af på kornstørrelsen - jo større korn, jo mere ligner egenskaberne egenskaberne for en enkelt krystal diamant," fortsatte Jalolov.
Forfatterne påpegede, at denne tilgang vil give mulighed for at studere de mekaniske egenskaber af ikke-enkeltkrystallinske materialer, der typisk syntetiseres og bruges i eksperimenter, samt at udføre omfattende undersøgelser af polykrystallinske og kompositmaterialer og opnå data så tæt på eksperimentelle resultater som muligt.
"I faktisk brug anvendes ofte materialer, der ikke er perfekte krystaller, på grund af deres manglende evne til perfekte krystaller til at opfylde kravene til et specifikt stykke udstyr fuldt ud."
"Et godt eksempel på dette er wolframcarbid og kobolt. Ved at tilføje kobolt til wolframcarbid bliver materialet mere revnebestandigt, hvilket gør det så værdifuldt i applikationer. Den nye metode vil give os mulighed for at undersøge årsagerne og måderne til at ændre det mekaniske egenskaber af disse flerfasesystemer på atomniveau," sagde Alexander Kvashnin, leder af forskningen og professor ved Energy Transition Center.
Flere oplysninger: Faridun N. Jalolov et al., Mechanical Properties of Single and Polycrystalline Solids from Machine Learning, Avanceret teori og simuleringer (2024). DOI:10.1002/adts.202301171
Leveret af Skolkovo Institute of Science and Technology
 Varme artikler
Varme artikler
-
 Fordelene ved rekonvalescent plasma til COVID-19 er stadig uklareDette scanningselektronmikroskopbillede viser SARS-CoV-2 (gul)-også kendt som 2019-nCoV, virussen, der forårsager COVID-19-isoleret fra en patient, der kommer fra overfladen af celler (blå/pink) dyr
Fordelene ved rekonvalescent plasma til COVID-19 er stadig uklareDette scanningselektronmikroskopbillede viser SARS-CoV-2 (gul)-også kendt som 2019-nCoV, virussen, der forårsager COVID-19-isoleret fra en patient, der kommer fra overfladen af celler (blå/pink) dyr -
 Mangan-enkeltatom-katalysator øger ydeevnen af elektrokemisk reduktion af kuldioxidSkematisk diagram af Mn SAC-fremstilling og mekanisme for elektrokemisk CO 2 reduktion. Kredit:FENG jiaqi Elektrokemisk CO 2 reduktionsreaktion (CO 2 RR) er en lovende tilgang til at omdanne
Mangan-enkeltatom-katalysator øger ydeevnen af elektrokemisk reduktion af kuldioxidSkematisk diagram af Mn SAC-fremstilling og mekanisme for elektrokemisk CO 2 reduktion. Kredit:FENG jiaqi Elektrokemisk CO 2 reduktionsreaktion (CO 2 RR) er en lovende tilgang til at omdanne -
 Realtidsdækning af hjernens indre muliggjortFig. 1 Forskelle mellem konventionel metabolomanalyse for dissekerede hjerneprøver og det nyudviklede in vivo realtidsovervågningssystem. Kredit:Nagoya University Overvågning i realtid af dynamikk
Realtidsdækning af hjernens indre muliggjortFig. 1 Forskelle mellem konventionel metabolomanalyse for dissekerede hjerneprøver og det nyudviklede in vivo realtidsovervågningssystem. Kredit:Nagoya University Overvågning i realtid af dynamikk -
 Meget selektivt adsorberende materiale tiltrækker uønskede materialer til gavn for biofremstillingEZ Select demonstrerer superhydrofobe egenskaber ved at afvise en dråbe vand. Kredit:Mary Kelly og Claire Kohout, Argonne National Laboratory En skummende stout, et solidt brød, en skarp ost; hver
Meget selektivt adsorberende materiale tiltrækker uønskede materialer til gavn for biofremstillingEZ Select demonstrerer superhydrofobe egenskaber ved at afvise en dråbe vand. Kredit:Mary Kelly og Claire Kohout, Argonne National Laboratory En skummende stout, et solidt brød, en skarp ost; hver
- Brug af mad til spilde i New York City
- Egypten placerer kolossen af Ramses II i atrium af nyt museum (Opdatering)
- Hvordan er en parallel kredsløb anderledes end en serie kredsløb?
- Forskere demonstrerer en teknisk tilgang til at kombinere medicin, kontrollere parasitære orme
- Kosmiske forstørrelsesglas giver et uafhængigt mål for universets ekspansion
- Studerer snesmeltningsepisoder af en pyrenæisk flod med et seismometer