


Forskere udvikler ny maskinlæringsmetode til modellering af kemiske reaktioner
Forskere fra Carnegie Mellon University og Los Alamos National Laboratory har brugt maskinlæring til at skabe en model, der kan simulere reaktive processer i en række forskellige organiske materialer og forhold.
"Det er et værktøj, der kan bruges til at undersøge flere reaktioner på dette felt," sagde Shuhao Zhang, en kandidatstuderende ved Carnegie Mellon Universitys afdeling for kemi. "Vi kan tilbyde en fuld simulering af reaktionsmekanismerne."
Zhang er den første forfatter på papiret, der forklarer skabelsen og resultaterne af denne nye maskinlæringsmodel med titlen "Exploring the Frontiers of Chemistry with a General Reactive Machine Learning Potential", udgivet i Nature Chemistry .
Selvom forskere har simuleret reaktioner før, havde tidligere metoder flere problemer. Reaktive kraftfeltmodeller er relativt almindelige, men de kræver normalt træning for specifikke reaktionstyper. Traditionelle modeller, der bruger kvantemekanik, hvor kemiske reaktioner simuleres baseret på underliggende fysik, kan anvendes på alle materialer og molekyler, men disse modeller kræver, at der bruges supercomputere.
Dette nye generelle interatomiske potentiale for maskinindlæring (ANI-1xnr) kan udføre simuleringer for vilkårlige materialer, der indeholder grundstofferne kulstof, brint, nitrogen og oxygen og kræver væsentlig mindre computerkraft og tid end traditionelle kvantemekaniske modeller. Ifølge Olexandr Isayev, lektor i kemi ved Carnegie Mellon og leder af laboratoriet, hvor modellen blev udviklet, skyldes dette gennembrud udviklingen inden for maskinlæring.
"Machine learning dukker op som en kraftfuld tilgang til at konstruere forskellige former for overførbare atomistiske potentialer ved hjælp af regressionsalgoritmer. Det overordnede mål med dette projekt er at udvikle en maskinlæringsmetode, der er i stand til at forudsige reaktionsenergier og -hastigheder for kemiske processer med høj nøjagtighed, men med en meget lav beregningsomkostning," sagde Isayev.
"Vi har vist, at disse maskinlæringsmodeller kan trænes på høje niveauer af kvantemekanikteori og med succes kan forudsige energier og kræfter med kvantemekanisk nøjagtighed og en stigning i hastigheden på så meget som 6-7 størrelsesordener. Dette er en ny paradigme i reaktive simuleringer."
Forskere testede ANI-1xnr på forskellige kemiske problemer, herunder sammenligning af biobrændstoftilsætningsstoffer og sporing af metanforbrænding. De genskabte endda Miller-eksperimentet, et berømt kemisk eksperiment, der skulle demonstrere, hvordan liv opstod på Jorden. Ved at bruge dette eksperiment fandt de ud af, at ANI-1xnr-modellen producerede nøjagtige resultater i kondenserede fasesystemer.
Zhang sagde, at modellen potentielt kunne bruges til andre områder inden for kemi med videreuddannelse.
"Vi fandt ud af, at det potentielt kan bruges til at simulere biokemiske processer som enzymatiske reaktioner," sagde Zhang. "Vi har ikke designet det til at blive brugt på en sådan måde, men efter modifikation kan det bruges til det formål."
I fremtiden planlægger teamet at forfine ANI-1xnr og lade det arbejde med flere grundstoffer og i flere kemiske områder, og de vil forsøge at øge omfanget af de reaktioner, det kan behandle. Dette kunne gøre det muligt at bruge det på flere områder, hvor design af nye kemiske reaktioner kunne være relevant, såsom opdagelse af lægemidler.
Zhang og Isayev fik selskab af Małgorzata Z. Makoś, Ryan B. Jadrich, Elfi Kraka, Kipton Barros, Benjamin T. Nebgen, Sergei Tretiak, Nicholas Lubbers, Richard A. Messerly og Justin S. Smith i denne undersøgelse.
Flere oplysninger: Udforskning af kemiens grænser med et generelt reaktivt maskinlæringspotentiale, Naturkemi (2024). DOI:10.1038/s41557-023-01427-3
Journaloplysninger: Naturkemi
Leveret af Carnegie Mellon University
 Varme artikler
Varme artikler
-
 Nyt mikroskop sætter rekord for visualisering af overfladebefugtningsegenskaberMikroskopets dråbesonde på en superhydrofob gylden fuglevinge (Troides aeacus) sommerfuglevinge. Kredit:Matti Hokkanen / Aalto University Mikroskopet er 1000 gange mere præcist end nuværende tekni
Nyt mikroskop sætter rekord for visualisering af overfladebefugtningsegenskaberMikroskopets dråbesonde på en superhydrofob gylden fuglevinge (Troides aeacus) sommerfuglevinge. Kredit:Matti Hokkanen / Aalto University Mikroskopet er 1000 gange mere præcist end nuværende tekni -
 En mere bæredygtig måde at raffinere metaller påStrategi for at reducere miljøbelastningen af en raffineringsproces:Erstat farlige kemikalier med mere godartede og genanvendelige forbindelser. Kredit:Michael J. Krause (Western University) Et
En mere bæredygtig måde at raffinere metaller påStrategi for at reducere miljøbelastningen af en raffineringsproces:Erstat farlige kemikalier med mere godartede og genanvendelige forbindelser. Kredit:Michael J. Krause (Western University) Et -
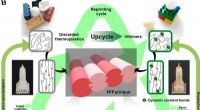 Upcycling af plast gennem dynamisk tværbindingCirkulær modeldesign af upcycling af termoplast til genanvendelig vitrimer til FFF. (A) Skematisk diagram, der sammenligner karakteristika for traditionelle termoplast, termohærdende og vitrimere. (B)
Upcycling af plast gennem dynamisk tværbindingCirkulær modeldesign af upcycling af termoplast til genanvendelig vitrimer til FFF. (A) Skematisk diagram, der sammenligner karakteristika for traditionelle termoplast, termohærdende og vitrimere. (B) -
 Dyrkning af 4-D-væv-den selvbøjede hornhindeForskere ved Newcastle University har udviklet et biologisk system, der lader celler danne en ønsket form ved at forme deres omgivende materiale-i første omgang skabe en selvbøjning af hornhinde. Hor
Dyrkning af 4-D-væv-den selvbøjede hornhindeForskere ved Newcastle University har udviklet et biologisk system, der lader celler danne en ønsket form ved at forme deres omgivende materiale-i første omgang skabe en selvbøjning af hornhinde. Hor
- Hvorfor er det forkert at sælge sin krop? Forstå liberale vs konservative moralske indvendinger mo…
- Koraller i grumset vand mindre påvirket af temperaturstress, forskningsfund
- Omkonfigurerbar elektronik viser løfte om bærbar, implanterbare enheder
- 1897 Lake Elmo UFO Encounter
- Kemikere udvikler en katalysator til at oxidere alkaner under milde forhold
- Hvad er taktiske atomvåben? Sikkerhedsekspert forklarer og vurderer, hvad de betyder for krigen i U…