


Beregningsforskere genererer molekylære datasæt i ekstrem skala
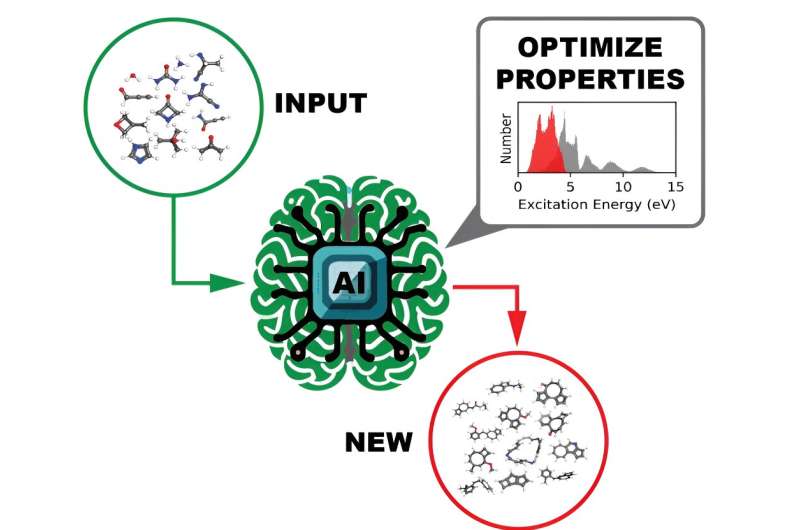
Et team af beregningsforskere ved Department of Energy's Oak Ridge National Laboratory har genereret og frigivet datasæt af hidtil uset skala, der giver de ultraviolette synlige spektrale egenskaber af over 10 millioner organiske molekyler. At forstå, hvordan et molekyle interagerer med lys, er afgørende for at afdække dets elektroniske og optiske egenskaber, som igen har potentielle fotoaktive anvendelser i produkter såsom solceller eller medicinske billedsystemer.
Ved at bruge højtydende computerressourcer på Oak Ridge Leadership Computing Facility kørte ORNL-teamet kvantekemiberegninger for at generere de enorme datasæt. For hvert af disse organiske molekyler kørte holdet atomistiske materialemodelleringsberegninger med forskellige tilnærmelser for at beregne forskellige ophidsede tilstandsegenskaber af interesse. Holdets resultater blev offentliggjort i Scientific Data .
Den ultimative tilsigtede anvendelse af open source-datasæt er at træne en dyb læringsmodel til at identificere molekyler med skræddersyede optoelektroniske og fotoreaktivitetsegenskaber, en tilgang, der er meget hurtigere og lettere at udføre end nuværende metoder.
"Brugen af DL-modeller til molekylært design er afgørende, fordi det kemiske rum, der skal udforskes for at søge efter disse molekyler, er ekstremt stort," sagde hovedforfatter Massimiliano Lupo Pasini, en dataforsker i ORNL's Computational Sciences and Engineering Division.
"Både eksperimenter og eksisterende første-principper-beregninger, som er baseret på de fysiske love, der bestemmer, hvordan stof og energi interagerer på det subatomare niveau, er simpelthen uoverkommelige af forskellige årsager. Eksperimenter er arbejdskrævende, og første-principper-beregninger kan nemt slå supercomputing til. Men DL-modeller giver meget lovende værktøjer til at overvinde disse barrierer," sagde Lupo Pasini.
Projektet kom i gang, da Stephan Irle, leder af ORNL's Computational Chemistry and Nanomaterials Sciences-gruppe, identificerede de ultraviolet-synlige spektrum af molekyler som en nyttig egenskab at forudsige med DL-modeller.
Opbygning af en DL-model, der er tilstrækkelig kompleks til at identificere ønskelige molekylære egenskaber, kræver træning af den med enorme mængder data, der udforsker alle forskellige områder af det kemiske rum. Jo flere data der indsamles, jo mere kan DL-modellen, der er trænet på den, opnå den nødvendige robusthed og generaliserbarhed for at fungere effektivt. Indsamling af så store mængder videnskabelige data til skalerbar DL kan dog give problemer med dataflow, især på faciliteter med flere brugere som OLCF, en DOE Office of Science-brugerfacilitet placeret på ORNL.
"En udfordring, der opstår, når der genereres store mængder data er, at antallet af filer, der skal administreres, stiger drastisk. Hvis det ikke administreres korrekt, kan en så stor mængde data kompromittere funktionen af det parallelle filsystem, som er en vigtig del af staten -of-the-art HPC-faciliteter," sagde Lupo Pasini.
For at løse denne udfordring samarbejdede Lupo Pasini med ORNL-datamatiker Kshitij Mehta om at udvikle en skalerbar workflow-software, der sikrer, at filerne genereret af kvantemekanikkoden håndteres korrekt uden at stresse filsystemet, såsom OLCF's Orion, som er en delt ressource, der håndterer input, output og lagring af data på supercomputersystemer.
Som en proof-of-concept-test genererede holdet GDB-9-Ex-datasættet med 96.766 molekyler sammensat af kulstof, nitrogen, oxygen og fluor med højst ni ikke-hydrogenatomer. Den viste, at den designede arbejdsgang er effektiv, og at DL-træningen præcist forudsiger positionen og intensiteten af de mest relevante toppe i det ultraviolet-synlige spektrum.
Fra den første succes øgede holdet sit volumen med ORNL_AISD-Ex-datasættet, som indeholder 10.502.917 molekyler sammensat af kulstof, nitrogen, oxygen, fluor og svovl med højst 71 ikke-brintatomer. Pilsun Yoo, en postdoktoral forskningsmedarbejder i Irles gruppe, udviklede værktøjer til at analysere de resulterende datasæt.
Det ultraviolet-synlige spektrum, som beskriver et molekyles excitationstilstande, blev beregnet for hvert af de mere end 10 millioner molekyler. Denne information afslører, hvilken lysfrekvens der kræves for at målrette et molekyle og bryde nogle bindinger af den kemiske forbindelse ad.
En anden egenskab af interesse beregnet for hvert molekyle var HOMO-LUMO-gabet - energigabet mellem den højest besatte molekylære orbital og den laveste ubesatte molekylære orbital - som pålideligt måler molekylets stabilitet. Med disse oplysninger kunne en DL-model effektivt gennemsøge dataene for at identificere lovende molekyler til forskellige mulige anvendelser.
Faktisk er Lupo Pasini og hans team hos ORNL, inklusive beregningsforsker i maskinlæring Pei Zhang og HPC-dataforsker Jong Youl Choi, ved at udvikle netop sådan en DL-model:HydraGNN.
"HydraGNN-arkitekturen tager den atomare struktur ind, konverterer den til en graf, og så forsøger den at forudsige som et output, hvad koden for første principper ville producere. Det er en surrogatmodel for dyre første-principper-beregninger," sagde Lupo Pasini.
Resultaterne fra HydraGNN's træning i datasættene og dets molekylære opdagelser vil blive beskrevet detaljeret i et kommende papir.
Flere oplysninger: Massimiliano Lupo Pasini et al., To datasæt med exciterede tilstande til kvantekemiske UV-vis spektre af organiske molekyler, Videnskabelige data (2023). DOI:10.1038/s41597-023-02408-4
Journaloplysninger: Videnskabelige data
Leveret af Oak Ridge National Laboratory
 Varme artikler
Varme artikler
-
 Forskere udvikler kryogen kaffekværnteknologiRegelmæssig (venstre) og kryogen (højre) slibning. Kredit:Coffeehouse Seneca Skoltech Ph.D. Dima Smirnov og hans kolleger fra St. Petersborg, Alexander Saichenko, Vladimir Dvortsov, Mikhail Tkache
Forskere udvikler kryogen kaffekværnteknologiRegelmæssig (venstre) og kryogen (højre) slibning. Kredit:Coffeehouse Seneca Skoltech Ph.D. Dima Smirnov og hans kolleger fra St. Petersborg, Alexander Saichenko, Vladimir Dvortsov, Mikhail Tkache -
 Fluorescensmønstre hjælper medicinsk diagnostikI den normale sekvens (ovenfor) bevæger elektronen sig langsomt og blinker langsomt. I tilfælde af en mutation bevæger elektronen sig hurtigt og blinker hurtigt. Hastigheden af elektronbevægelse mål
Fluorescensmønstre hjælper medicinsk diagnostikI den normale sekvens (ovenfor) bevæger elektronen sig langsomt og blinker langsomt. I tilfælde af en mutation bevæger elektronen sig hurtigt og blinker hurtigt. Hastigheden af elektronbevægelse mål -
 Følger kemikaliernes vej gennem jordenForskere fra Aarhus Universitet har udviklet en forbedret metode til at følge transporten af kemikalier gennem jorden. Kredit:Janne Hansen Hvor bliver pesticider og deres nedbrydningsprodukter a
Følger kemikaliernes vej gennem jordenForskere fra Aarhus Universitet har udviklet en forbedret metode til at følge transporten af kemikalier gennem jorden. Kredit:Janne Hansen Hvor bliver pesticider og deres nedbrydningsprodukter a -
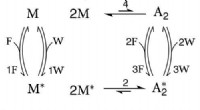 Ny termodynamisk ramme til cellerModel for energilagring og drevet syntese. Uden (hv. med) den orange stiplede overgang, det kemiske reaktionsnetværk modellerer energilagring (hhv. drevet syntese). Den højenergiske art A2A2 er i lav
Ny termodynamisk ramme til cellerModel for energilagring og drevet syntese. Uden (hv. med) den orange stiplede overgang, det kemiske reaktionsnetværk modellerer energilagring (hhv. drevet syntese). Den højenergiske art A2A2 er i lav
- Undersøgelsen afslører, at brasilianere ønsker at være tættere på naturen
- Journalister, som Twitter bruger, viser, at de taler i mindre bobler
- Apple sænker udsigterne, ser udfordringer i Kina, nye markeder
- Eksperiment og teori forenes endelig i debat om mikrobielle nanotråde
- Nye kort over asteroiden Psyche afslører en gammel verden af metal og rock
- El-scootere:Elsker eller hader du dem? Her er hvad du behøver at vide