


Hvordan man dyrker et evolutionært træ
Samle data :Indsaml DNA- eller proteinsekvensdata fra en forskelligartet gruppe af organismer, inklusive organismerne af interesse og nogle udgrupper til sammenligning.
Multiple Sequence Alignment :Juster sekvenserne for at skabe en multipel sekvensjustering. Denne justering bør overveje rækkefølgen og ligheden af nukleotider eller aminosyrer.
Vælg en træbygningsmetode: Vælg en fylogenetisk træbygningsmetode, såsom Maximum Likelihood, Neighbor-Joining eller Bayesiansk inferens. Hver metode bruger forskellige algoritmer og statistiske tilgange til at beregne evolutionære sammenhænge.
Konstruer træet :Brug den valgte metode til at konstruere det evolutionære træ. Outputtet vil normalt være et diagram eller kladogram, der repræsenterer forgreningsmønstrene og evolutionære forhold mellem de undersøgte organismer.
Bootstrap-analyse (valgfrit) :Udfør bootstrapping for at vurdere den statistiske støtte for forgreningsmønstrene i træet. Bootstrap-analyse omsampler gentagne gange dataene for at skabe flere træer, hvilket giver statistiske konfidensværdier (dvs. bootstrap-værdier) for hver gren.
Root træet :Identificer og angiv roden af træet. Dette er typisk en forfædres knude eller en udgruppe, der repræsenterer den seneste fælles forfader af alle organismerne i træet.
Fortolk træet :Analyser forgreningsmønstrene og længderne for at udlede evolutionære forhold, divergenstider og potentielle fælles forfædre. Identificer klader (grupper af organismer, der deler en fælles forfader) og den undersøgte arts evolutionære historie.
Juster træet (valgfrit) :Hvis yderligere data bliver tilgængelige, eller der bruges forskellige parametre, kan træet forfines eller opdateres for at forbedre dets nøjagtighed og opløsning.
Sidste artikelHvorfor var der så mange dinosaurarter?
Næste artikelHvad tænder afslører om moderne menneskers liv
 Varme artikler
Varme artikler
-
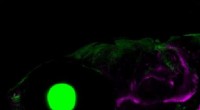 Hvordan udviklede hvirveldyr først kæber?En zebrafisk, der viser skelettet og kæben (magenta), øjet (grøn cirkel på leT) og gællelignende pseudogren og gæller (grønne strukturer til højre). Kredit:Mathi Thiruppathy/Crump Lab For fem hundr
Hvordan udviklede hvirveldyr først kæber?En zebrafisk, der viser skelettet og kæben (magenta), øjet (grøn cirkel på leT) og gællelignende pseudogren og gæller (grønne strukturer til højre). Kredit:Mathi Thiruppathy/Crump Lab For fem hundr -
 Brug af kemisk fingeraftryk til at bekæmpe svindel med fisk og skaldyr og ulovligt fiskeriFisk og skaldyr er et af de mest handlede fødevareprodukter i verden. Kredit:Saya Kimura/Pexels, CC BY Falske fødevarer invaderer vores supermarkeder, da fødevarer, vi elsker, erstattes eller forfa
Brug af kemisk fingeraftryk til at bekæmpe svindel med fisk og skaldyr og ulovligt fiskeriFisk og skaldyr er et af de mest handlede fødevareprodukter i verden. Kredit:Saya Kimura/Pexels, CC BY Falske fødevarer invaderer vores supermarkeder, da fødevarer, vi elsker, erstattes eller forfa -
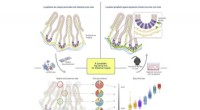 Hvordan tarmen erstatter og reparerer sig selvGrafisk abstrakt. Kredit:Cell stamcelle (2022). DOI:10.1016/j.stem.2022.05.007 For at fungere som en robust barriere mod patogener og samtidig absorbere nødvendige næringsstoffer, skal slimhinden i
Hvordan tarmen erstatter og reparerer sig selvGrafisk abstrakt. Kredit:Cell stamcelle (2022). DOI:10.1016/j.stem.2022.05.007 For at fungere som en robust barriere mod patogener og samtidig absorbere nødvendige næringsstoffer, skal slimhinden i -
 Stop og gå i kaliumkanalenFysikeren arbejder med meget grundlæggende kaliumkanaler for ikke at gøre analysen unødigt kompliceret. Kredit:Katrin Binner Celler har brug for åbninger i cellemembranen for at kunne foretage udv
Stop og gå i kaliumkanalenFysikeren arbejder med meget grundlæggende kaliumkanaler for ikke at gøre analysen unødigt kompliceret. Kredit:Katrin Binner Celler har brug for åbninger i cellemembranen for at kunne foretage udv
- Klimaændringernes mønstre
- Hvordan hærmyrernes ikoniske masseangreb udviklede sig
- Forbedret fokus for kontormedarbejdere
- ALMA opdager koldt støv omkring den nærmeste stjerne
- Ydeevnen forbedres, når fjendens fjende er en ven
- Hvordan hjernen genkender ansigter:Maskinlæringssystem gengiver spontant aspekter af menneskelig ne…