


Formbaseret model kaster lys over forenklet proteinbinding
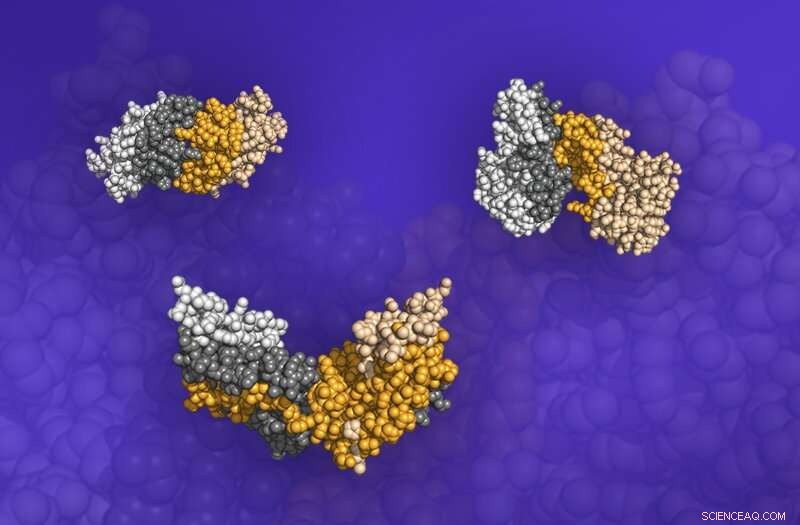
Tre dimerer, proteinstrukturer bestående af to bundne proteiner, fra Dockground-databasen. Grænsefladerne, hvor proteinerne mødes, vises som de mørkede områder. Kredit:ORNL
Kan noget så simpelt som form fuldt ud afgøre, om proteiner vil binde sammen eller ej? Forskere idriftsætter supercomputere for at finde ud af det.
Et hold ledet af Sharon Glotzer, fremtrædende professor og afdelingsformand for kemiteknik ved University of Michigan (UM), brugte 200 petaflop-supercomputeren ved det amerikanske energiministeriums (DOE's) Oak Ridge National Laboratory (ORNL) til at modellere låse-og-nøgle-interaktioner mellem proteiner for at studere deres bindingsadfærd. Resultaterne, udgivet i Soft Matter, afslørede, at nogle proteiner gør, faktisk, binde baseret på form alene.
"Vi har demonstreret, at noget så simpelt som form er i stand til at forudsige proteininteraktioner, der undertiden er virkelig komplekse, sagde Jens Glaser, computational scientist i Advanced Computing for Chemistry and Materials-gruppen ved Oak Ridge Leadership Computing Facility (OLCF). "Denne første demonstration har fået os til at tro, at form har været en upåskønnet ingrediens i mange proteinsamlingsprocesser."
Resultaterne kan have mange anvendelser i biologisk forskning. For eksempel, metoden kan bruges til at screene medicin mod sygdomme eller give forskere oplysninger om, hvordan man bruger proteiner som byggesten til at designe nye biologiske materialer.
"Denne spændende undersøgelse viser styrken af formkomplementaritet i forudsigelsen af protein-protein-grænseflader, " sagde Dr. Stephanie McElhinny, programleder ved US Army Combat Capabilities Development Command's Army Research Laboratory, henviser til det gunstige rumlige forhold mellem to kompatibelt formede proteiner. "Beregningsmodeller, der præcist forudsiger disse grænseflader, vil understøtte det fremtidige design af avancerede proteinbaserede materialer med aktive og responsive egenskaber, såsom lys-høstende proteinbaseret plast, der kunne fungere som et kunstigt blad til elproduktion."
Supercomputere afslører, at form er nøglen i nogle proteiner
For at proteiner med succes kan binde til hinanden, en af dem fungerer som en ligand, et molekyle, der binder sig til et målprotein, og en af dem fungerer som en receptor, molekylet, der modtager liganden. Denne proces involverer komplekse kemiske interaktioner, hvor molekyler deler bindinger og ændrer deres konfigurationer ved binding.
Glotzers team ønskede at se, om de kunne forudsige denne molekylære binding baseret på form alene, ignorerer interaktionen mellem proteiner. Fra en database med mere end 6, 000 proteinpar, holdet testede 46 par, der vides at binde sig til hinanden, og simulerede deres samling på Summit. Holdet udførte simuleringerne under INCITE-programmet (Innovative and Novel Computational Impact on Theory and Experiment).
Ligesom flere tennisbolde bliver kastet mod et enkelt mål, simuleringerne modellerede flere ligander, der blev kastet på en enkelt, fast målreceptor. Ud af de 46 testede par, de fandt 6 par, der klarede sig godt - mere end 50 procent af den tid, de lykkedes at samle baseret udelukkende på deres komplementære former.
"Vi kiggede på grænsefladerne, hvor proteinerne bandt sammen for at se, hvor ens de lignede deres virkelige grænseflader, og så bestemte vi cutoff for at se, hvor mange par der var gode forudsigere for de rigtige grænseflader, " sagde Fengyi Gao, Ph.D. kandidat ved UM. "Vi fandt ud af, at 13 procent af disse proteinpar kunne binde baseret på form alene."
Holdet byggede derefter en maskinlæringsmodel, der kunne bestemme, hvilke proteiner der er i stand til at samle udelukkende baseret på form. At kombinere deres oprindelige model med sådanne maskinlæringsværktøjer vil hjælpe dem med at forstå, hvilken information der er nødvendig for proteinpar, der ikke kan samles alene baseret på formkomplementaritet.
Kører proteiner parallelt
At modellere flere reversible bindingsprocesser af 46 proteinpar under forskellige parametre, de havde brug for to dages beregningstid og mere end 3, 000 GPU'er - et beløb, som kun en supercomputer som OLCF's Summit kunne levere. OLCF er et DOE Office of Science User Facility på ORNL.
Som en del af den HOOMD-blå beregningskode, der blev brugt til at køre simuleringerne, Glaser, som tidligere var assisterende forsker i Glotzers gruppe ved UM, udviklet en algoritme, der simulerede proteinerne i nærvær af mange små partikler. Men Glaser fandt en måde kun at modellere bevægelsen af de proteiner, holdet var interesseret i, undgå unødvendige og dyre beregninger for opløsningsmiddelmolekylerne omkring dem.
"Jeg kørte koden parallelt, så mange forskellige parametre, iterationer af det samme system, og forskellige proteiner kunne fordeles på tværs af GPU'erne, "Sagde Glaser." Dette gjorde det let for os at gøre brug af Summits parallelle computermuligheder. "
Ved hjælp af Summit, holdet fangede seks proteinpar, der kun bandt baseret på formkomplementaritet, hvor en af dem opnår binding mere end 94 procent af tiden.
"Det var ganske overraskende for os, at en sådan forenklet model korrekt kunne vælge netop den ene pose, som de antager ud af de mange hundrede eller flere stillinger, der konkurrerer, " sagde Glaser. "Vi forventede, at meget mere ville være nødvendigt for at reproducere den reelle bindingsposition for disse proteinpar."
Modeller kan hjælpe med lægemiddelscreening
Holdet planlægger at studere flere proteiner, der også kan binde baseret på form - eller danne endnu højere ordensstrukturer. Teamets nuværende undersøgelse undersøgte kun proteindimerer, som består af to proteiner bundet sammen, men teamet ønsker at kende begrænsningen for, hvordan proteinformer kan udvikle sig til at danne hierarkiske proteinstrukturer.
"Før vi lavede denne undersøgelse, Jeg forventede faktisk ikke, at proteiner kunne danne dimerer baseret på form alene, " sagde Fengyi Gao, Ph.D. kandidat ved UM. "Men nu, vi har fundet ud af, at dette virker, og vi kan studere mere komplekse strukturer eller endda kombinere dette med andre tilgange, som maskinlæring, for at se, hvilke funktioner vi skal bruge for at aktivere den korrekte binding."
Teamet håber, at de til sidst kan forudsige bindingen af protein-protein-grænseflader i proteinklynger eller proteinkrystalliseringsstrukturer.
"Vi tror, vi kan tilpasse denne tilgang til noget som lægemiddelscreening i fremtiden, " sagde Gao. "Ud over det, Vi håber, at denne formbaserede model kan tjene som grundlag for at studere proteinsamling generelt."
 Varme artikler
Varme artikler
-
 Team tager et stort skridt mod trykte anisotrope magneterBilleder a) og b) viser mikrografer af additiv fremstillingsbundet magnet, efter udskrivning, men før justering. Figur c) viser et mikrograf af magneten efter justering, og figur d) viser røntgendiffr
Team tager et stort skridt mod trykte anisotrope magneterBilleder a) og b) viser mikrografer af additiv fremstillingsbundet magnet, efter udskrivning, men før justering. Figur c) viser et mikrograf af magneten efter justering, og figur d) viser røntgendiffr -
 Hvordan evolution bygger de mest effektive flyvepladerSvømmere og løbesedler kan nedbrydes til trykproducerende (orange) og trækproducerende (blå) dele, med fremdriften passende repræsenteret af et oscillerende flyveblad. Kredit:(c) Procedurer fra Natio
Hvordan evolution bygger de mest effektive flyvepladerSvømmere og løbesedler kan nedbrydes til trykproducerende (orange) og trækproducerende (blå) dele, med fremdriften passende repræsenteret af et oscillerende flyveblad. Kredit:(c) Procedurer fra Natio -
 Opsætning af grundlæggende baser for informationsmetasurfaces(a) Et sæt målesteder i k-rum. (b) Teoretiske intensitetsfordelinger af målepunkterne. (c, d) En prøve af 1-bit uordnet fasemønster af metasoverfladen og den genererede fjernfelts intensitetsfordeling
Opsætning af grundlæggende baser for informationsmetasurfaces(a) Et sæt målesteder i k-rum. (b) Teoretiske intensitetsfordelinger af målepunkterne. (c, d) En prøve af 1-bit uordnet fasemønster af metasoverfladen og den genererede fjernfelts intensitetsfordeling -
 Underlig superleder fører dobbelt livEn usædvanlig egenskab ved superledende materialer er, at de udviser magnetfelter og dermed får magneter til at svæve, som vist her. En undersøgelse på SLAC og Stanford af en særligt underlig superled
Underlig superleder fører dobbelt livEn usædvanlig egenskab ved superledende materialer er, at de udviser magnetfelter og dermed får magneter til at svæve, som vist her. En undersøgelse på SLAC og Stanford af en særligt underlig superled
- Hvordan sårbare grupper blev efterladt i pandemirespons
- Guld nanopartikelkæder begrænser lyset til nanoskalaen
- Hvorfor kvindelige ledere udmærker sig under coronavirus-pandemien
- Hvor indflydelsesrige er fysisk geologi på mennesker?
- Frygt for klimaændringer driver forskere ud af laboratoriet og ud på gaderne
- Hvordan klapperslangeskæl hjælper dem med at nippe til regnvand fra deres kroppe