


Fujitsu udvikler molekylær simuleringsteknologi til effektivt at skabe nye lægemiddelkandidater

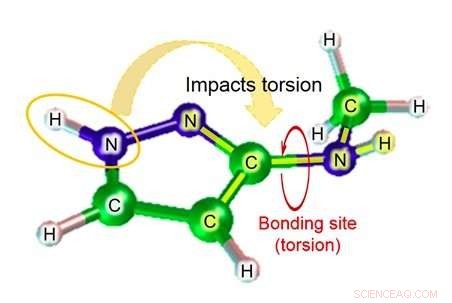
Figur 1:Dihedral vinkel (vinklen dannet af planet skabt af atomer A, B, og C, og flyet skabt af atomer B, C, og D). Kredit:Fujitsu
Fujitsu Laboratories annoncerede i dag udviklingen af molekylær simuleringsteknologi til opdagelse af lægemidler, der præcist kan estimere bindingsaffinitet, hvilket repræsenterer i hvilken grad proteiner, der kan forårsage sygdomme (målproteiner), binder sig til kemiske stoffer, der kan blive kandidatlægemidler. I processen med at opdage medicin, der er et krav om nøjagtig forudsigelse af bindingsaffiniteten mellem målproteiner og kemiske stoffer, som giver et groft skøn over et lægemiddels effekt. Molekylær simuleringsteknologi har tidligere været meget udbredt som en metode til at forudsige bindingsaffinitet, beregning af de omtrentlige kræfter, der opstår mellem atomer i molekyler ved hjælp af newtonsk mekanik. Problemet med denne metode, imidlertid, forbliver, at den lave gradnøjagtighed af dets estimering af de vigtigste parametre-graden af torsion på bindingsstederne. Dette betyder, at nøjagtigheden af dets estimering af den samlede bindingsaffinitet også er dårlig.
Nu, Fujitsu Laboratories har udviklet molekylær simuleringsteknologi, der estimerer vridningsgraden i et kemisk stof, som er direkte forbundet med den forudsagte bindingsaffinitet. Den nye teknologi tager ikke kun hensyn til bindingsstedet, hvor torsionen vil forekomme, men også virkningen af nærliggende atomer. Fujitsu Laboratories vurderede denne teknologi til 190 typer kemiske stoffer, sammenligning af resultaterne med korrekte resultater, der blev opnået fra første principberegning og derefter evaluering af fejlprocenten. Ved at gøre det, det var i stand til at bekræfte, at fejlprocenten i estimatet af torsionsgraden var, gennemsnitlig, en tiendedel af tidligere teknologi. Det forventes, at brugen af denne nye teknologi til it-baseret lægemiddelfund, med sin evne til nøjagtigt at estimere bindingsaffiniteten for målrettede proteiner og kemiske stoffer, giver mulighed for banebrydende nye lægemiddelopdagelsesbestræbelser, der ikke kunne opnås med tidligere tilgange.
Opdagelsen af nye lægemidler kræver betydelige udgifter og tidsrammer, der kan måles i årtier, fører til en global søgning efter nye metoder til at opdage medicin. En af de metoder, der har fået stor interesse, er it-baseret lægemiddelfund, en ny metode til opdagelse af lægemidler ved hjælp af computere, der gør det muligt at skabe kemiske stoffer som kandidater til nye lægemidler med stor sandsynlighed for succes. IT-baseret lægemiddelfund er blevet et omdrejningspunkt for forventninger som en banebrydende teknologi til oprettelse af nye lægemidler, fordi i modsætning til tidligere metoder til forsøg og fejl, hvor kemiske stoffer gentagne gange skabes og testes, denne fremgangsmåde gør det muligt praktisk talt at designe kemiske stoffer og vurdere deres virkninger.
Figur 2:Eksempel på molekylær struktur:3- (methylamino) pyrazol. Kredit:Fujitsu
Virkningen af et kemisk stof som et lægemiddel udtrykkes, når det kemiske stof binder sig til et målprotein. Når det kemiske stof binder sig til målproteinet, det kan ændre sin form på linje med målproteinets. Graden af deformation, nemlig, parametrene, der angiver omfanget af denne formændring, er direkte forbundet med stoffets og proteinets bindingsaffinitet, og giver en grov ide om dens virkning som et lægemiddel. I betragtning af dette, der er en stærk efterspørgsel efter evnen til præcist at forudsige denne værdi. For at beregne deformationsgraden af et kemisk stof, der er metoder baseret på kvantemekanik og metoder baseret på newtonsk mekanik. Kvantemekanikbaseret beregning af første principper muliggør ekstremt nøjagtige beregninger, at løse elektronernes tilstande fra de involverede atomers typer og positioner. På den anden side, imidlertid, evnen hos de første principper til at udføre krævende beregninger nødvendigvis fører til massiv tid, der kræves for at fuldføre beregningerne. For at simulere graden af deformation for talrige kemiske stoffer, den nødvendige tid er i størrelsesordenen år, gør denne metode upraktisk. På den anden side, omtrentlige beregninger baseret på molekylære simuleringer er ekstremt hurtige, ved hjælp af newtonsk mekanik til at beregne kræfterne mellem atomerne i molekylerne, og kan endda nemt håndtere store molekyler som proteiner. Følgelig, denne metode er meget udbredt. Med Newtonian mekanik, kræfterne mellem atomerne udtrykkes på følgende måde:
- Som en kraft, der afhænger af afstanden mellem to atomer bundet til hinanden
- En kraft, der er afhængig af vinklerne mellem tre atomer bundet til hinanden
- En kraft, der er afhængig af graden af torsion i bindingen, og
- En kraft, der er afhængig af afstanden mellem atomer, der ikke er bundet.
Imellem disse, når et kemisk stof er bundet til et målprotein, graden af torsion af bindingen repræsenterer den vigtige grad af deformation. Med eksisterende teknologi, imidlertid, nøjagtigheden af estimeringen af parameteren dihedral vinkel (figur 1), som er nødvendig for at beregne graden af torsion af bindingen, er ret lav, hvilket resulterer i problemet med lav nøjagtighed i estimeringen af affiniteten af bindingen i simuleringen.
Fujitsu Laboratories har udviklet molekylær simuleringsteknologi i mere end ti år. Nu, ved hjælp af den viden, den opnåede ved tidligere bestræbelser, Fujitsu Laboratories har udviklet en molekylær simuleringsteknologi, der kan estimere dihedralvinkelparameteren ved at tage hensyn til virkningen af atomer nær bindingen. Eksisterende teknologi estimerer parameteren dihedral vinkel baseret på i alt fire atomer-de to atomer i den relevante binding, og de andre atomer hver af disse atomer var bundet til. Afhængigt af molekylets struktur, imidlertid, der er tilfælde, hvor atomer ud over de fire kan have en betydelig indvirkning, og i disse tilfælde, estimatets fejlmargin kan være ret stor. Med denne teknologi, Fujitsu Laboratories har oprettet en database med estimeringsformler til delvise strukturmønstre, hvor virkningen af atomer længere væk fra bindingsstedet kan være betydelig, såvel som for graden af torsion af kemiske stoffer, der ville kunne forventes i så fald. Ved hjælp af den relevante estimeringsformel til at finde graden af torsion (figur 2) for molekyler, der svarer til databasen for delstrukturer, det er blevet muligt selv at foretage meget nøjagtige estimater for molekylær torsion, hvilket tidligere var svært at beregne nøjagtigt.
Da Fujitsu Laboratories integrerede denne teknologi i den software, den havde udviklet til at generere sofistikerede parametre for kræfterne mellem atomer (FF-FOM), det var i stand til at bekræfte, at resultaterne var i overensstemmelse med nøjagtige beregninger.

Figur 3:Evaluering af ydelsen af dihedrale vinkelparameterværdier ved hjælp af 190 typer kemiske sammensatte strukturer. Kredit:Fujitsu
Da Fujitsu Laboratories vurderede forskellen mellem resultaterne af denne teknologi og resultaterne af en beregning fra de første principper til estimering af torsionsgraden med 190 typer kemiske stoffer, det var mindre end en tiendedel af den tidligere teknologi, gennemsnitlig, 0,6 kcal/mol under termiske udsving ved stuetemperatur, bekræfter, at den nye teknologi er praktisk. Fordi det præcist kan estimere bindingsaffiniteten for målproteiner og kemiske stoffer, det forventes, at brugen af denne teknologi vil føre til skabelse af banebrydende nye lægemidler gennem dens anvendelse i it-baseret lægemiddelfund.
 Varme artikler
Varme artikler
-
 pH-værdien af calciumioner kontrollerer ionkanalåbningenEn dørvogterring:Det ekstracytosoliske/lumenale domæne af den menneskelige TRPML2-ionkanal, hvis struktur denne undersøgelse belyste for første gang, fungerer som en pH-afhængig calciumregulator. Det
pH-værdien af calciumioner kontrollerer ionkanalåbningenEn dørvogterring:Det ekstracytosoliske/lumenale domæne af den menneskelige TRPML2-ionkanal, hvis struktur denne undersøgelse belyste for første gang, fungerer som en pH-afhængig calciumregulator. Det -
 Smeltepunkter af metaller vs. ikke-metallerEt elements smeltepunkt er, når det konverteres fra fast form til en væske. Metaller, som er fysisk fleksible elementer, der kan lede varme og elektricitet, har en tendens til at være faste ved stuete
Smeltepunkter af metaller vs. ikke-metallerEt elements smeltepunkt er, når det konverteres fra fast form til en væske. Metaller, som er fysisk fleksible elementer, der kan lede varme og elektricitet, har en tendens til at være faste ved stuete -
 Ledende natur i krystalstrukturer afsløret ved forstørrelse på 10 millioner gangeUniversity of Minnesota Professor K. Andre Mkhoyan og hans team brugte analytisk scanning transmissionselektronmikroskopi (STEM), som kombinerer billeddannelse med spektroskopi, at observere metallisk
Ledende natur i krystalstrukturer afsløret ved forstørrelse på 10 millioner gangeUniversity of Minnesota Professor K. Andre Mkhoyan og hans team brugte analytisk scanning transmissionselektronmikroskopi (STEM), som kombinerer billeddannelse med spektroskopi, at observere metallisk -
 Forskere konstruerer hårdere mikrober for at forbedre bioproduktion af brændstoffer, kemikalierForskere arbejder på at identificere og producere robuste enzymer, der kan erstatte andre enzymer, der nedbrydes i gæringen af biobaserede brændstoffer og kemikalier. Kredit:Laura Jarboe. Travl,
Forskere konstruerer hårdere mikrober for at forbedre bioproduktion af brændstoffer, kemikalierForskere arbejder på at identificere og producere robuste enzymer, der kan erstatte andre enzymer, der nedbrydes i gæringen af biobaserede brændstoffer og kemikalier. Kredit:Laura Jarboe. Travl,
- Undersøgelse afslører nye muligheder for at hjælpe virksomheder med at forbedre processen for til…
- Brug af TikTok som et pædagogisk værktøj til kropssprog kan forbedre elevernes læring
- Apples medstifter protesterer på Facebook ved at lukke en konto
- Skift de klimafaktorer, der styrer vegetationsdynamikken på det tibetanske plateau
- Fransk tomatavler tager imod Monsanto over ukrudtsmiddel
- Undersøgelse undersøger de bedste ledelsesmåder for iværksættere