


Forskere afkoder dynamikken i den største proteinnedbrydende maskine i atomare detaljer

Kredit:CC0 Public Domain
Protein nanomaskiner lavet af flere proteinmolekyler er meget dynamiske under deres handlinger på deres funktionelle mål, nogle gange kaldet substrater. Dynamikken af disse store protein nanomaskiner med mere end megadalton molekylvægt er modstandsdygtige over for strukturel analyse af eksisterende teknologi som røntgenkrystallografi og nuklear magnetisk resonansspektroskopi. Kryo-elektronmikroskopi (cryo-EM), en ny teknologi til strukturbestemmelse i høj opløsning, har potentiale til at visualisere dynamikken i store protein nanomaskiner, men de eksisterende cryo-EM-rekonstruktioner af meget dynamiske strukturer er blevet begrænset til moderat til lav opløsning.
Forskere har længe drømt om at afkode dynamikken i store molekylære maskiner af megadalton-størrelser i atomare detaljer, den ultimative determinant for deres biologiske funktioner. Nu, et hold af biofysikere fra Peking Universitet, Dana-Farber Cancer Institute og Harvard Medical School har brugt cryo-EM til at visualisere dynamikken på atomniveau af 2,5 megadalton proteasomet, den største kendte proteinnedbrydende maskine i eukaryote celler, under dets kemo-mekaniske virkning på et proteinsubstrat. De rekonstruerede en næsten komplet dynamisk procedure for substratbehandling i det menneskelige proteasom med hidtil uset opløsning, der tillod bestemmelsen af atomare detaljer i 3-D, beslægtet med at filme en 3D-film atom for atom.
"Dette arbejde baner vejen for at studere termodynamik af megadalton nanomaskiner med atompræcision langt væk fra ligevægt, " sagde Youdong Mao, en biofysiker og tilsvarende forfatter på et nyt banebrydende papir offentliggjort i det første nummer af tidsskriftet Natur i 2019. "Dette studie åbner op for adskillige muligheder for strukturbaseret lægemiddelopdagelse rettet mod humant proteasom til behandling af myelomatose og neurodegenerative sygdomme."
Ubiquitin-proteasomsystemet (UPS) er den vigtigste proteinnedbrydningsvej i celler. Det opretholder balancen mellem proteinmaterialer i levende celler, og spiller en afgørende rolle i hurtig nedbrydning af regulatoriske proteiner, fejlfoldede proteiner eller beskadigede proteiner. UPS er involveret i formentlig alle cellulære processer, såsom cellecyklus, regulering af genekspression og så videre. Unormal proteinmetabolisme forårsaget af UPS-lidelse er direkte relateret til mange menneskelige sygdomme, herunder kræft. I 2004 Aaron Ciechanover, Irwin Rose og Avram Hershko blev tildelt Nobelprisen i kemi for deres opdagelse af denne nedbrydningsvej. I hjertet af UPS er det proteasom, der er ansvarligt for nedbrydning af ubiquitin-mærkede substrater. Det er en af de mest fundamentale og komplicerede gigantiske holoenzymmaskiner i celler. Humant proteasom holoenzym indeholder mindst 33 forskellige underenhedstyper med en samlet molekylvægt på ca. 2,5 megadalton. Det er også kendt som det direkte mål for adskillige småmolekylære lægemidler godkendt af FDA i USA til behandling af myelomatose.
Brug af cryo-EM i kombination med maskinlæringsteknologi, holdet bestemte dynamiske strukturer af det substrat-engagerede humane proteasom i syv mellemliggende konformationelle tilstande ved 2,8-3,6 Å opløsning, fanget under nedbrydning af et polyubiquityleret protein. Ved denne beslutning, holdet var i stand til at identificere enkelte magnesiumioner bundet til både ATP og ADP i cryo-EM-densitetskortene. Disse 3-D strukturer belyser et bemærkelsesværdigt rumligt kontinuum af dynamiske substrat-proteasom-interaktioner.
Spændende nok, holdet fandt, at initieringen af substrattranslokation i vid udstrækning er koordineret med andre dynamiske regulatoriske begivenheder, der forbereder proteasomet til processiv substratnedbrydning. Gennem yderligere systematisk analyse, holdet opdagede, hvordan den kemiske energi af ATP-hydrolyse omdannes til det mekaniske arbejde med substrat, der udfolder sig gennem en meget samordnet proces af multi-protein konformationelle ændringer.
Deres fund giver ny indsigt i den komplette cyklus af substratbehandling og foreslår forskellige måder efterfulgt af ATP-hydrolyse i proteasom-holoenzymet. Det menes at være første gang, at en komplet cyklus af sekventiel ATP-hydrolyse i en AAA-ATPase heterohexamermotor blev visualiseret på atomniveau. Dette løser en langvarig videnskabelig debat om ATPase-hexamerer mellem to hypotesemodeller, den ene tyder på sekventiel ATP-hydrolyse og den anden antager tilfældige hydrolytiske hændelser i den hexamere ring. Især holdet observerede tre hovedformer for højt koordineret ATP-hydrolyse, med hydrolytiske hændelser i to modsat placerede ATPaser, i to tilstødende ATPaser, og i én ATPase ad gangen. Disse hydrolytiske tilstande regulerer elegant deubiquitylering, initiering af translokation, og processiv udfoldning af substrater, henholdsvis.
Holdet bemærkede visse begrænsninger i denne undersøgelse, herunder at mangfoldigheden af nukleotidbehandlingshændelser i distinkte ATPaser under overgange mellem på hinanden følgende tilstande af proteasomet kan have resulteret i fravær af hurtige trin og tyndt befolkede mellemtilstande i deres kryo-EM-rekonstruktioner. Holdet forestiller sig udsigten til yderligere udforskninger i denne henseende ved at identificere disse manglende mellemled for at afklare, hvordan ATP-hydrolytiske hændelser og nukleotidudveksling koordineres med hinanden, og allosterisk forbundet med substrattranslokation. "Yderligere udvikling inden for dataanalyseteknologi er nødvendig for at udtrække endnu mere dynamisk information fra det samme datasæt, " sagde Mao. "Der er lang vej at gå for datadrevet maskinlæringsteknologi til fuldt ud at frigøre den potentielle kraft af cryo-EM til at løse komplekse dynamikker i megadalton molekylære maskiner."
Sidste artikelPartnere i katalyse:En effektiv vej til umættede ketoner
Næste artikelStruktur af fedtforarbejdningsenzym bestemt
 Varme artikler
Varme artikler
-
 Ansigtsmaskeindsats kan hjælpe med at diagnosticere sygdomstilstande, viser undersøgelseKredit:Pixabay/CC0 Public Domain På baggrund af aktuelle begivenheder, mange mennesker bærer ansigtsmasker for at beskytte sig selv og andre. Men den samme ansigtsmaske kunne en dag også indsamle
Ansigtsmaskeindsats kan hjælpe med at diagnosticere sygdomstilstande, viser undersøgelseKredit:Pixabay/CC0 Public Domain På baggrund af aktuelle begivenheder, mange mennesker bærer ansigtsmasker for at beskytte sig selv og andre. Men den samme ansigtsmaske kunne en dag også indsamle -
 Forskere bruger origami til at løse rumrejseudfordringerForskerne har udviklet en origami-inspireret, foldet plastbrændstofblære, der ikke revner ved superkolde temperaturer og en dag kan bruges til at opbevare og pumpe brændstof. Kredit:WSU WSU-forske
Forskere bruger origami til at løse rumrejseudfordringerForskerne har udviklet en origami-inspireret, foldet plastbrændstofblære, der ikke revner ved superkolde temperaturer og en dag kan bruges til at opbevare og pumpe brændstof. Kredit:WSU WSU-forske -
 Nye fotokatalysatorer kan udføre solcelledrevet omdannelse af kuldioxid til brændstofVedtagelse af den fotokatalytiske omdannelse af CO2 til brændstof i højemissionsanlæg ville være yderst gavnlig for både miljøet og økonomien. Kredit:Shutterstock Forskere ved Daegu Gyeongbuk Inst
Nye fotokatalysatorer kan udføre solcelledrevet omdannelse af kuldioxid til brændstofVedtagelse af den fotokatalytiske omdannelse af CO2 til brændstof i højemissionsanlæg ville være yderst gavnlig for både miljøet og økonomien. Kredit:Shutterstock Forskere ved Daegu Gyeongbuk Inst -
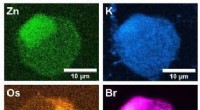 Forskere observerer, hvordan potentiel kræftbehandling reagerer i en enkelt celleRøntgenfluorescensbilleder taget ved I14-strålelinjen ved Diamond Light Source:Det grønne er zink, blå er kalium, orange er osmium og pink er brom. Kredit:University of Warwick Ved hjælp af en 185
Forskere observerer, hvordan potentiel kræftbehandling reagerer i en enkelt celleRøntgenfluorescensbilleder taget ved I14-strålelinjen ved Diamond Light Source:Det grønne er zink, blå er kalium, orange er osmium og pink er brom. Kredit:University of Warwick Ved hjælp af en 185
- 30% af Californiens jord skal bevares under ny ordre fra Gov. Newsoms
- Abiotiske faktorer i kystområderne
- Økonomisk opsving forhindrede flere robotter i at fjerne arbejdspladser
- Græsplæner til bælgfrugter:Minnesota betaler husejere for at plante bee græsplæner
- Rumæniens skove under stigende trussel - sammen med rangers
- Sådan konverteres gram til molekyler