


Ny software bringer cryo-EM-kort i lavere opløsning i fokus
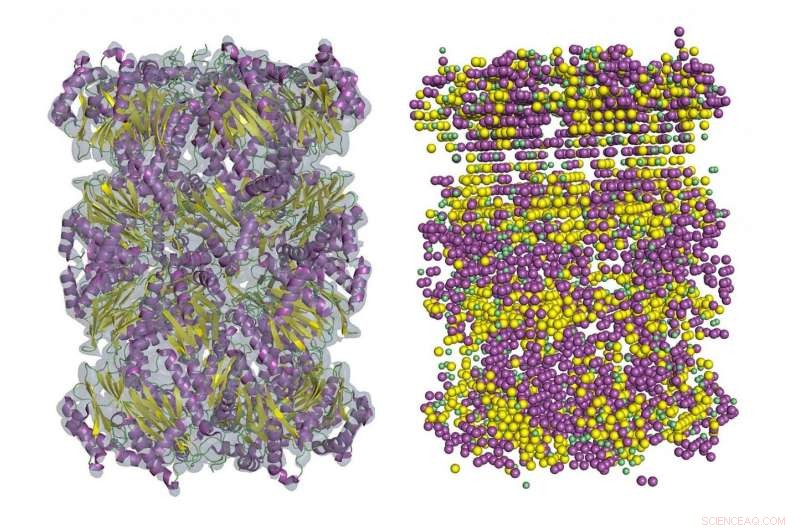
Et eksempel på den sekundære strukturdetektering i cryo-EM-densitetskort ved hjælp af Emap2Sec. Til venstre er et EM-kort over archaeal 20S proteasom (EMDB ID:EMD-1733). Til højre er detekteret sekundære strukturer af Emap2Sec. Punkter i magenta er positionerne af detekterede alfa-helixer; gule punkter er detekteret beta-strenge, og grønne punkter er for detekterede spoler (andre strukturer). Kredit:Purdue University billede/Daisuke Kihara
Kryo-elektronmikroskopi er nu den mest populære metode til at bestemme proteinstrukturer, som hjælper forskere med at udvikle lægemidler til forskellige slags lidelser. I løbet af de sidste mange årtier, det har erstattet røntgenkrystallografi, fordi det kan afbilde proteiner, der ikke let kan formes til store krystaller. Den nye teknik var så revolutionerende, at den vandt sine udviklere Nobelprisen i kemi i 2017.
Det endelige produkt af cryo-EM er et kort over tætheden af atomer i biologiske molekyler, men for at opnå det detaljeringsniveau, forskerne har brug for, de skal foretage yderligere analyser. En ny undersøgelse i tidsskriftet Naturens metoder skitserer en teknik til at bringe lavopløsningskort op til par.
Den tilgang, forskerne bruger til at gøre dette, afhænger af detaljeringsgraden, de starter med. Kort ved 2 til 3 ångström (Å, en længdeenhed, der bruges til at udtrykke størrelsen af atomer og molekyler) betragtes generelt som højopløselige. Imidlertid, kort af denne kvalitet er svære at opnå, og mange produceres stadig almindeligvis i intervallet 4 til 10 Å. Af alle proteiner deponeret til elektronmikroskopidatabanken fra 2016-18, mere end 50 % blev løst ved mellemopløsning.
"Hvis opløsningen er bedre end tre, så kan konventionelle værktøjer spore aminosyreposition og bygge et kort over atompositioner. Men ofte kan cryo-EM ikke give dig et 3 Å kort, " sagde Daisuke Kihara, en professor i biologiske videnskaber og datalogi ved Purdue University. "På kort på 5 Å eller lavere, du kan normalt slet ikke se kædeforbindelser."
Proteiner er faktisk kæder af aminosyrer, og binding mellem aminogrupper og carboxylgrupper skaber nogle gange visse foldningsmønstre. Disse mønstre, kendt som alfa-helixer og beta-strenge, danner proteinets sekundære struktur.
På kort fra 5 til 8 Å, nogle fragmenter af den sekundære struktur af proteiner er normalt synlige, men det ville være meget svært at spore hele kæden. Kiharas nye metode, kendt som Emap2sec, afdækker sekundære strukturer i kort fra 6 til 10 Å.
Emap2sec har et dybt foldet neuralt netværk i kernen af sin algoritme. Disse netværk er deep-learning-systemer, der primært bruges til at klassificere billeder, gruppere dem efter lighed og udføre objektgenkendelse. Det virker til proteinstrukturidentifikation i 3-D-kort, fordi metoden "konvolverer" lokale korttæthedstræk til billeder af en større region, når informationen passerer gennem lag af neurale netværk. Den lokale forudsigelse er lavet i sammenhæng med et stort område af kortet.
Identificerede sekundære strukturer i 3-D-kort hjælper forskere med at tildele kendte strukturer af proteiner, der allerede er blevet løst i kortet. Det betyder, at de nogle gange har et udgangspunkt, eller i det mindste et fingerpeg om, hvordan noget af strukturen ser ud. Emap2sec kan hjælpe forskere med at passe deres brik ind i puslespillet hurtigere og lettere. De identificerede strukturoplysninger kan også være nyttige til at finde fejl i strukturmodellering.
 Varme artikler
Varme artikler
-
 Dobbelts katalysator muliggør høj omdannelse af syngas til flydende carbonhydrider i benzininterva…Skematisk diagram for omdannelsen af syngas til flydende carbonhydrider i benzinområdet over en dobbeltbedskatalysator (CZA+Al2O3)/N-ZSM-5(97) og resultaterne af stabilitetstesten. Kredit:DICP B
Dobbelts katalysator muliggør høj omdannelse af syngas til flydende carbonhydrider i benzininterva…Skematisk diagram for omdannelsen af syngas til flydende carbonhydrider i benzinområdet over en dobbeltbedskatalysator (CZA+Al2O3)/N-ZSM-5(97) og resultaterne af stabilitetstesten. Kredit:DICP B -
 West Virginia forskere bruger neutroner til at studere materialer til forbedringer af kraftværkerForskere fra West Virginia University brugte VULCAN ved Spallation Neutron Source til at studere materialer kaldet højentropioxider til at udvikle industrielle og forbrugerbaserede applikationer til f
West Virginia forskere bruger neutroner til at studere materialer til forbedringer af kraftværkerForskere fra West Virginia University brugte VULCAN ved Spallation Neutron Source til at studere materialer kaldet højentropioxider til at udvikle industrielle og forbrugerbaserede applikationer til f -
 Marshmallow-lignende silikonegeler brugt som isolering i beholdere til kryokonserverede embryonerFoto af containeren, der pakker en MG. Kredit:Gen Hayase Da den genetiske modifikation af mus er mere og mere udbredt i medicinsk og biologisk forskning, så, også, er behovet for en effektiv måde
Marshmallow-lignende silikonegeler brugt som isolering i beholdere til kryokonserverede embryonerFoto af containeren, der pakker en MG. Kredit:Gen Hayase Da den genetiske modifikation af mus er mere og mere udbredt i medicinsk og biologisk forskning, så, også, er behovet for en effektiv måde -
 Vi har opdaget en måde at genvinde DNA fra fingeraftryk uden at ødelægge demKredit:Shutterstock Fingeraftryk indeholder meget mere information, end du måske er klar over. De giver ikke kun et mønster, som man kan identificere folk med. De kan også indeholde DNA. Og da hve
Vi har opdaget en måde at genvinde DNA fra fingeraftryk uden at ødelægge demKredit:Shutterstock Fingeraftryk indeholder meget mere information, end du måske er klar over. De giver ikke kun et mønster, som man kan identificere folk med. De kan også indeholde DNA. Og da hve
- Forskning afslører Kinas vende emissionsstrømme
- Forskere opnår den højeste certificerede effektivitet af organiske solceller til dato
- Ny cellulær billeddannelse baner vej for kræftbehandling
- Bezos er ikke berørt af konkurrenceproblemer på Amazon
- Math Madness Svarark
- NASA-undersøgelse forudsiger mindre Sahara-støv i fremtidige vinde