


Molekylære simuleringer viser, hvordan lægemidler blokerer nøglereceptorer

Kredit:CC0 Public Domain
Mange lægemidler virker ved at målrette det, der er kendt som 'G-protein-koblede receptorer'. I en ny undersøgelse, forskere fra Uppsala Universitet beskriver, hvordan de har været i stand til at forudsige, hvordan specielle molekyler, der kan bruges i ny immunterapi mod kræft, binder til disse receptorer. Forskernes beregningsmetoder, præsenteret i journalen Angewandte Chemie er et vigtigt bidrag til fremtidens strukturbaserede lægemiddeldesign.
G-protein-koblede receptorer (GPCR'er) er blandt de proteinmålgrupper, der har størst betydning for lægemiddeludvikling. Disse receptorer reagerer på, for eksempel, lys, smag, lugter, adrenalin, histamin, dopamin og en lang liste over andre molekyler ved at transmittere yderligere biokemiske signaler inde i celler. Forskerne, der udførte undersøgelsen af GPCR'er, blev belønnet med Nobelprisen i kemi i 2012.
I dag, omkring 30 procent af alle lægemidler på markedet har GPCR'er som deres målproteiner. Nogle lægemiddelmolekyler, såsom morfin, aktiverer receptorerne (agonister), mens andre, såsom betablokkere, inaktivere dem (antagonister).
En vigtig GPCR er adenosin A2A -receptoren. Dets antagonister kan bruges i ny immunterapi mod cancer. Sammen med det biofarmaceutiske firma Sosei-Heptares, forskerne Willem Jespers, Det er lykkedes Johan Åqvist og Hugo Gutierrez-de-Terán fra Uppsala Universitet at vise, hvordan en række A2A-antagonister binder til receptoren og inaktiverer den.
Med molekylær dynamiske simuleringer og beregning af bindingsenergier, blev det muligt at forudsige, hvordan molekyler fra medicinalvirksomheden ville binde sig til receptorerne, og hvor stærkt de gør det. Derefter, nye antagonister blev designet, og syntetiseret af kemikere fra Santiago de Compostela University, Spanien. Tredimensionelle strukturer af komplekserne, der dannes mellem disse molekyler og receptoren, blev derefter bestemt eksperimentelt med røntgenkrystallografi. Computerberegninger viste sig i stand til at forudsige både strukturen og bindingsstyrken i komplekserne med høj præcision.
"Dette er et solidt skridt fremad, og det lykkedes os med stor præcision at forudsige, hvordan denne familie af molekyler binder A2A -receptoren. Vores beregningsmetoder har nu et stort gennembrud inden for strukturbaseret lægemiddeldesign, " siger Hugo Gutierrez-de-Terán, der stod i spidsen for Uppsala-gruppens projekt.
 Varme artikler
Varme artikler
-
 Forskere bliver ru med nanomaterialer for at eliminere problematisk klæbrighed forårsaget af glatt…Øverste billede (fra venstre mod højre):Undergraduate Katerina Kimes (siddende), Professor Tevis Jacobs, Bachelorstuderende Cameron Kisailus, og ph.d. Kandidat Abhijeet Gujrati ser på et kort over ove
Forskere bliver ru med nanomaterialer for at eliminere problematisk klæbrighed forårsaget af glatt…Øverste billede (fra venstre mod højre):Undergraduate Katerina Kimes (siddende), Professor Tevis Jacobs, Bachelorstuderende Cameron Kisailus, og ph.d. Kandidat Abhijeet Gujrati ser på et kort over ove -
 Nye materialer af perovskit udfordrer traditionelle forestillinger om højtrykskemiKemisk intuition fortæller os, at tryk har en tendens til at øge antallet af koordinering, og derfor normalt laver en struktur ordnet, der især gælder for perovskit-lignende forbindelser. Imidlertid,
Nye materialer af perovskit udfordrer traditionelle forestillinger om højtrykskemiKemisk intuition fortæller os, at tryk har en tendens til at øge antallet af koordinering, og derfor normalt laver en struktur ordnet, der især gælder for perovskit-lignende forbindelser. Imidlertid, -
 Lysstyrede polymerer kan skifte mellem robuste og blødeDesign af polyMOCer med fotoskiftbar topologi. Kredit:(c) Natur (2018). DOI:10.1038/s41586-018-0339-0 MIT -forskere har designet et polymermateriale, der kan ændre dets struktur som reaktion på
Lysstyrede polymerer kan skifte mellem robuste og blødeDesign af polyMOCer med fotoskiftbar topologi. Kredit:(c) Natur (2018). DOI:10.1038/s41586-018-0339-0 MIT -forskere har designet et polymermateriale, der kan ændre dets struktur som reaktion på -
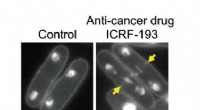 Anticancer-mekanisme afsløret i gæreksperimenterMed DNA topoisomerase II (topo II) intakt, DNA i fissionsgærceller replikeres normalt og deles jævnt mellem to datterceller (venstre). Når det er under virkningen af anti-cancer lægemidlet ICRF-193,
Anticancer-mekanisme afsløret i gæreksperimenterMed DNA topoisomerase II (topo II) intakt, DNA i fissionsgærceller replikeres normalt og deles jævnt mellem to datterceller (venstre). Når det er under virkningen af anti-cancer lægemidlet ICRF-193,
- Ormearter mistede 7, 000 gener efter at have udviklet sig til at befrugte sig selv
- Trio, der boede på rumstationen, vender sikkert tilbage til Jorden
- Design af selektive membraner til batterier ved hjælp af en værktøjskasse til opdagelse af lægem…
- Pas på hype - videnskabsspin udbredt, advarer forskere
- Navigerer i overgangen til ren energi under COVID-19-krisen
- Astronomer opdager flimring fra stjernen EF Aquilae