


Forskere på forkant med udvikling af maskinlæringsmetoder til kemisk opdagelse

Kredit:CC0 Public Domain
Opdagelsen og formuleringen af nye lægemidler, antivirale midler, antibiotika og generelt kemikalier med skræddersyede egenskaber er en lang og omhyggelig proces. Tværfaglig forskning i krydsfeltet mellem biokemi, fysik og datalogi kan ændre dette. Udvikling af machine learning (ML) metoder, kombineret med de første principper inden for kvante- og statistisk mekanik og trænet på stadig mere tilgængelige molekylære store datasæt, har potentialet til at revolutionere processen med kemisk opdagelse.
"Kemisk opdagelse og maskinlæring er bundet til at udvikle sig sammen, men at opnå ægte synergi mellem dem kræver løsning af mange enestående udfordringer, " siger Alexandre Tkatchenko, Professor i teoretisk kemisk fysik ved universitetet.
Maskinlæring for at hjælpe med at identificere lægemiddelkandidater
Universitetet indledte et samarbejde med den belgiske virksomhed Janssen Pharmaceuticals i foråret 2020 for at udvikle nye ML-metoder til at identificere forbindelser, der har et stærkt terapeutisk potentiale (også kaldet lægemiddelkandidater). Indtil nu, ML-tilgange er blevet udviklet til små molekyler. Dette forskningsprojekt har til formål at udvide arkitekturen og overførbarheden af kvantemekanik-baserede maskinlæringstilgange til store molekyler af farmaceutisk betydning.
"Genereringen af nye kemikalier med aktivitet på relevante biologiske mål er medicinalvirksomhedernes kerneforretning. Maskinlæringstilgange har potentiale til at fremskynde processen og reducere fejlrater i lægemiddelopdagelse. Efter at være blevet kontaktet af en førende medicinalvirksomhed for at arbejde sammen at identificere lægemiddelkandidater er et glædeligt tegn på den industrielle anerkendelse af vores ekspertise, " kommenterer Dr. Leonardo Medrano-Sandonas, en postdoktor i prof. Tkatchenkos gruppe.
Partner i et Innovative Training Network finansieret af Europa-Kommissionen
Sammen med tre store europæiske medicinalvirksomheder (Bayer, AstraZeneca, Janssen), kemivirksomheden Enamine og ti akademiske partnere med ekspertise inden for computerdesign af lægemidler, Prof. Tkatchenko har fået tildelt Marie Sklodowska-Curie Actions—Innovative Training Network-bevillingen til projektet Advanced machine learning for Innovative Drug Discovery (AIDD) for perioden 2021-2023. Dette projekt har til formål at udvikle innovative ML-metoder til at bidrage til en integreret "One Chemistry"-model, der kan forudsige resultater lige fra molekylegenerering til syntese og forstå, hvordan man sammenfletter kemi og biologi for at udvikle nye lægemidler.
Her slår den videnskabelige ekspertise sig sammen med de industrielle partneres medicinske og syntetiske kemiske ekspertise, og drager fordel af store værdifulde datasæt. For første gang, al metodologisk udvikling vil være tilgængelig open source. Træningsnetværket vil forberede en generation af forskere, der har færdigheder både i maskinlæring og kemi til at fremme medicinsk kemi.
"At lave præcise forudsigelser ved at bruge maskinlæring kritisk afhænger af adgang til store samlinger af højkvalitetsdata og domæneekspertise til at analysere dem, " forklarer prof. Tkatchenko. "At sætte vores kræfter sammen er et første skridt mod en kemisk opdagelsesrevolution drevet af maskinlæring."
Området for maskinlæring til kemisk opdagelse er ved at dukke op, og betydelige fremskridt forventes at ske i den nærmeste fremtid. Prof. Tkatchenko har for nylig publiceret en artikel i tidsskriftet Naturkommunikation hvor han diskuterer de seneste gennembrud på dette område og fremhæver udfordringerne for de kommende år. Artiklen er tilgængelig online.
 Varme artikler
Varme artikler
-
 Krystalstruktur afslører, hvordan curcumin hæmmer kræftEt 3D billede, opnået ved hjælp af røntgenkrystallografi, viser curcumin i gul og rød binding til kinase-enzym dual-specificity tyrosin-reguleret kinase 2 (DYRK2) i hvidt på atomniveau. Kredit:UC San
Krystalstruktur afslører, hvordan curcumin hæmmer kræftEt 3D billede, opnået ved hjælp af røntgenkrystallografi, viser curcumin i gul og rød binding til kinase-enzym dual-specificity tyrosin-reguleret kinase 2 (DYRK2) i hvidt på atomniveau. Kredit:UC San -
 Forskning kunne minimere uønskede bivirkninger i nye lægemidlerKredit:CC0 Public Domain Opioider lindrer smerter, men kan forårsage respirationssvigt og død ved overdosering. Antipsykotiske lægemidler kan hjælpe mennesker med at klare psykiske sygdomme, men m
Forskning kunne minimere uønskede bivirkninger i nye lægemidlerKredit:CC0 Public Domain Opioider lindrer smerter, men kan forårsage respirationssvigt og død ved overdosering. Antipsykotiske lægemidler kan hjælpe mennesker med at klare psykiske sygdomme, men m -
 Forskere rapporterer 3-D-printet latexgummi gennembrudEn tværfaglig gruppe af forskere i kemi og maskinteknik udviklede en ny proces til 3D-print af latexgummi. Latex gummidele, såsom dette pumpehjul printet ved 100 mikron opløsning, tillade ikke-destruk
Forskere rapporterer 3-D-printet latexgummi gennembrudEn tværfaglig gruppe af forskere i kemi og maskinteknik udviklede en ny proces til 3D-print af latexgummi. Latex gummidele, såsom dette pumpehjul printet ved 100 mikron opløsning, tillade ikke-destruk -
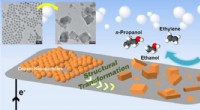 Kobberkatalysator giver højeffektiv CO2-til-brændstofkonverteringSkematisk af en ny katalysator lavet af kobbernanopartikler, der omdanner kuldioxid til multicarbonprodukter (ethylen, ethanol, og propanol). Øverst til venstre er transmissionselektronmikroskopbilled
Kobberkatalysator giver højeffektiv CO2-til-brændstofkonverteringSkematisk af en ny katalysator lavet af kobbernanopartikler, der omdanner kuldioxid til multicarbonprodukter (ethylen, ethanol, og propanol). Øverst til venstre er transmissionselektronmikroskopbilled
- Et skridt tættere på en datamotorvej til fremtidens internet
- Black Friday bliver en skygge af sit tidligere jeg i USA
- Lysosom: Definition, struktur og funktion
- Forskere dyrker kunstige hår med et smart fysiktrick
- I krigsramte Gaza, vandforurening bag sundhedsproblemer
- En af de tætteste galaksehobe i universet afsløres