


Brug af en radikal til at bryde C-F-bindinger en ad gangen
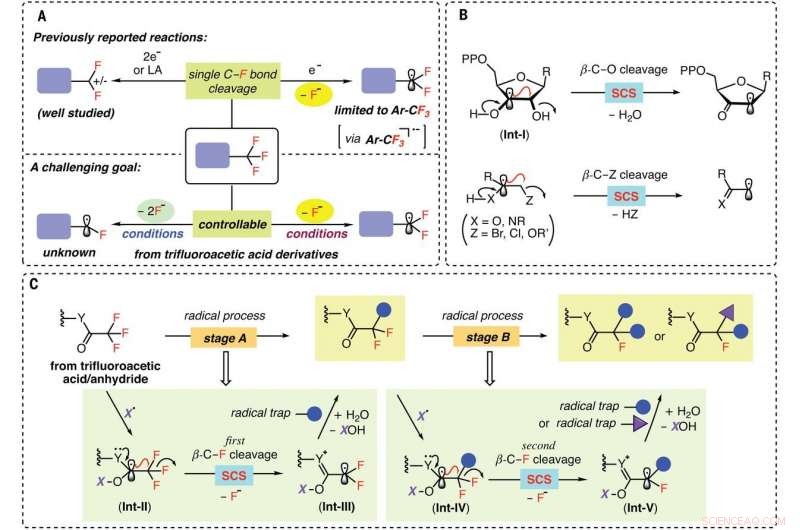
Strategier for C-F-bindingsfunktionaliseringer af CF3-grupper. (A) Defluorering af CF3-grupper til difluorsubstituerede mellemprodukter (øverst) og det udfordrende mål om kontrollerbart at generere di- og monofluoralkylradikaler (nederst). (B) Spin-center shift (SCS) i biokemiske omdannelser og organisk syntese. (C) Dette arbejde:To-trins proces til sekventiel C–F-bindingsspaltning via SCS. Kredit: Videnskab (2021). DOI:10.1126/science.abg0781
Et team af forskere fra University of Science and Technology i Kina og University of California har fundet en måde at bruge radikaler til at bryde C-F-bindinger en ad gangen, når de arbejder med trifluoracetamider og acetater. I deres papir offentliggjort i tidsskriftet Videnskab , gruppen beskriver, hvordan de fandt det rigtige radikal til sådanne reaktioner, og hvordan deres teknik kan bruges i fremtidige applikationer.
Kemikere har søgt efter måder at tilføje fluor til visse lægemidler, fordi det hjælper deres molekyler med at bevæge sig gennem cellemembraner. Problemet har været at finde en måde at udskifte blot et af fluoratomerne i forbindelser som trifluormethyl for at skabe mono- og/eller difluorerede forbindelser. Med de nuværende metoder, når en reaktion bryder den første C-F-binding, de to andre bliver svagere, resulterer i deres fjernelse, såvel. I denne nye indsats, forskerne har fundet en måde at udføre sådanne reaktioner på uden at svække sekundære C-F-bindinger.
Forskerne hævder, at løsningen på problemet ligger i at finde den rigtige radikal - en, der kan målrette trifluoracetat og trifluoracetamid udgangsmaterialer. Efter en omfattende søgning, de fandt ud af, at amino-borylgruppen ville målrette mod udgangsforbindelser som ønsket. Den radikale arbejdede, de bemærker, fordi CF 3 kulstofmolekyle var stærkt elektronmangel. Det betød, at når den radikale brød den første C-F-binding, det var meget mindre sandsynligt at gøre det samme for den anden eller tredje C-F-obligation. Slutresultatet var en reaktion, der stoppede, før alle fluorbindingerne var blevet brudt.
Holdet testede deres radikal sammen med en aminoboran ved at bytte en C-F-binding ud med enten en CH-binding eller en C-C-binding, samtidig med at man tilføjer andre ingredienser såsom brint for at lave en lang række mono- og difluorerede acetamider og acetater. De brugte også deres radikal til at modificere eksisterende lægemiddelmolekyler for at gøre dem mere effektive til at målrette mod specifikt væv. De foreslår, at deres teknik skulle vise sig at være nyttig i udviklingen af nye terapier for en lang række lidelser, lige fra lymfom til endometriose.
© 2021 Science X Network
 Varme artikler
Varme artikler
-
 Højhastigheds atomkraftmikroskopi visualiserer celleproteinfabrikkerModel til oversættelse af ribosomer og forlængelsesfaktorer. EF1A • GTP • aatRNA og EF2 samles til den ribosomale stilk på det translaterende ribosom. Oversættelsesfaktorpuljen bidrager til effektiv p
Højhastigheds atomkraftmikroskopi visualiserer celleproteinfabrikkerModel til oversættelse af ribosomer og forlængelsesfaktorer. EF1A • GTP • aatRNA og EF2 samles til den ribosomale stilk på det translaterende ribosom. Oversættelsesfaktorpuljen bidrager til effektiv p -
 Forskere tager et fingerpeg fra naturen om at skabe skudsikre belægningerKredit:University of Houston Reje, hummer og svampe virker måske ikke som gode redskaber til slagmarken, men tre ingeniører fra University of Houston bruger kitin-et derivat af glukose, der findes
Forskere tager et fingerpeg fra naturen om at skabe skudsikre belægningerKredit:University of Houston Reje, hummer og svampe virker måske ikke som gode redskaber til slagmarken, men tre ingeniører fra University of Houston bruger kitin-et derivat af glukose, der findes -
 Meget elastisk biologisk nedbrydelig hydrogel til bioprint af nyt vævYi Hong, UTA professor i bioteknik og leder af projektet. Kredit:UTA Forskere ved University of Texas i Arlington har udviklet en meget elastisk biologisk nedbrydelig hydrogel til bio-printning af
Meget elastisk biologisk nedbrydelig hydrogel til bioprint af nyt vævYi Hong, UTA professor i bioteknik og leder af projektet. Kredit:UTA Forskere ved University of Texas i Arlington har udviklet en meget elastisk biologisk nedbrydelig hydrogel til bio-printning af -
 Ny strategi til fremstilling af enkeltatom-katalysatorer via elektrokemisk aflejringKredit:CC0 Public Domain Forskere fra University of Science and Technology of China (USTC) fra det kinesiske videnskabsakademi (CAS) har rapporteret en strategi for at fremstille single-atom katal
Ny strategi til fremstilling af enkeltatom-katalysatorer via elektrokemisk aflejringKredit:CC0 Public Domain Forskere fra University of Science and Technology of China (USTC) fra det kinesiske videnskabsakademi (CAS) har rapporteret en strategi for at fremstille single-atom katal
- UCF sælger eksperimentelt Martian snavs - $ 20 per kilo, plus forsendelse
- Astronomer opdager en ultra-højenergi gammastrålekilde
- Hvad er kvantemærkelighed?
- Engelsktalende er mere tilbøjelige til at modtage rådgivning om automatisk selvmordsforebyggelse
- Tesla -aktier falder efter 3Q -leverancer savner estimater
- 10 typer fysisk forandring