


Forskning afdækker manglende fysik i eksplosive hotspots
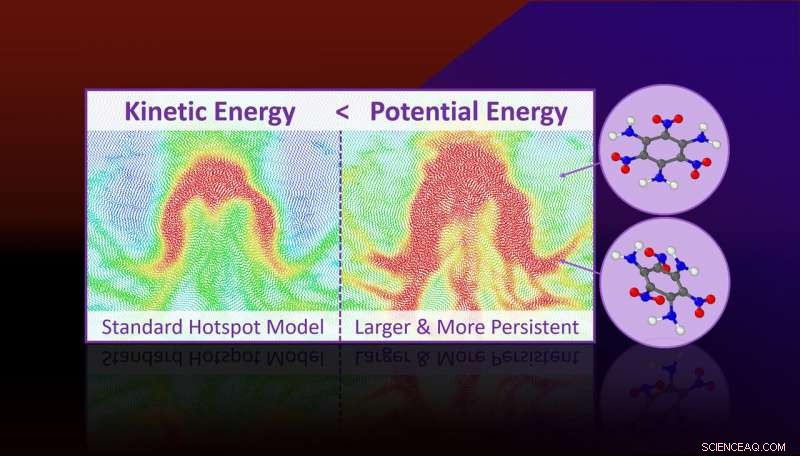
Molekylær dynamik simuleringer forudsiger, at mere potentiel energi er lokaliseret i hotspots, end deres kinetiske energi (eller temperatur) antyder. Overskydende potentiel energi er bundet til vedvarende anstrengte molekylære tilstande, der er klaret til kemiske reaktioner og forklarer, hvorfor hotspots reagerer hurtigere end hovedparten. Kredit:Lawrence Livermore National Laboratory
Forskning udført på Lawrence Livermore National Laboratorys (LLNL) supercomputer Quartz fremhæver resultater foretaget af forskere, der afslører et manglende aspekt af fysikken i hotspots i TATB (1, 3, 5-trimamino-2, 4, 6-trinitrobenzen) og andre sprængstoffer.
Hotspots er lokaliserede områder med forhøjet temperatur, der dannes fra stød-induceret kollaps af mikrostrukturel porøsitet og er kendt for at styre stødinitierings- og detonationsegenskaberne af eksplosiver. Hovedkonceptet bag hotspots er, at lokale forhøjede temperaturer accelererer lokal kemi.
Forskningen er omtalt i 11. marts-udgaven af Journal of Physical Chemistry Letters og var et samarbejde mellem LLNL og Purdue University. Forfattere inkluderer Matthew Kroonblawd fra LLNL og Brenden Hamilton, Chunyu Li og Alejandro Strachan fra Purdue.
Arbejdet fremhæver et forsømt fysisk aspekt af de tidlige stadier af hotspot-dannelse og -evolution, der giver en rute til systematisk at forbedre multifysiske modeller for stødinitiering og detonation, der bruges til at vurdere ydeevne og sikkerhed.
"Et af de mest forvirrende resultater fra tidlige reaktive simuleringer af molekylær dynamik er, at hotspots dannet ved sammenbrudte porer reagerer meget hurtigere end dem med tilsvarende størrelse, temperatur og tryk i bulkmaterialet, " sagde Strachan. "Mens han blev genkendt, årsagen bag disse forskelle blev ikke forstået. Vores undersøgelse løser dette spørgsmål ved, at vi finder ud af, at det eksplosive materiale i en kollapset pore er fundamentalt forskelligt fra hovedmassen, og at det er i en højenergitilstand, der er forberedt til kemiske reaktioner."
Vigtigheden af at forstå hotspots
TATB er et ufølsomt højeksplosiv, der er kritisk for landets nukleare lager og er udfordrende at modellere på kontinuumskalaen. Tekniske modeller for eksplosiv sikkerhed og detonationsydelse er afhængige af fysikmodeller, der fokuserer på dannelsen og væksten af hotspots.
Kroonblawd forklarede, at "multifysiske modeller på kontinuumniveau, der bruges til at vurdere sikkerhed og ydeevne, er meget empiriske, hvilket gør det vanskeligt at skabe eksplosive modeller, der kan overføres til forskellige anvendelsesforhold. Manglen på overførbare modeller gælder især for ufølsomme højeksplosive stoffer såsom TATB. Det er stadig ikke muligt at bygge en eksplosiv model ud fra de første principper, hvilket indikerer, at nøgleaspekter mangler i vores forståelse af hotspotfysik og kemi."
Disse modeller er afhængige af nøjagtige behandlinger af kemisk reaktivitet og termisk transport; hvorvidt hotspots vil vokse og samle sig til en detonationsbølge, bestemmes af en konkurrence mellem varmegenereringshastigheden på grund af kemi og varmetab på grund af varmeledning.
At identificere årsagen bag forskelle i hotspot-reaktionshastigheder giver en vej til at formulere mere generelle eksplosive modeller, der vil forbedre deres forudsigelige nøjagtighed og overførbarhed. Mens disse modeller typisk har fokuseret på temperatur som den vigtigste variabel, der kontrollerer kemi, resultaterne tyder på, at omarbejdning af disse modeller med hensyn til den potentielle energi vil give en mere generel behandling, der kan skelne de forskellige reaktiviteter i forskellige materielle tilstande.
Gennem simuleringer af molekylær dynamik med alle atomer, forskerne fandt ud af, at hotspots ikke kun er områder med lokaliseret kinetisk energi (eller temperatur), men er også regioner med lokaliseret potentiel energi. Mængden af potentiel energi er meget større end mængden af kinetisk energi, og den er koncentreret i molekylære tilstande, der er relevante for kemisk nedbrydning.
Den potentielle energilokalisering manifesterer sig på grund af belastninger på molekylært niveau i plastisk deformerede områder af materialet, og dette vil føre til en mekanokemisk acceleration af reaktioner.
"Nøglen er, at der ikke er et en-til-en forhold mellem kinetisk og potentiel energi i disse systemer, derfor, man kan ikke udlede lokale reaktionshastigheder udelukkende fra temperaturfeltet, " sagde Hamilton.
Teamet udfører store simuleringer
Arbejdet, udført af Materials Science Divisions personale i LLNL Energetic Materials Center (EMC) og Materials Engineering Department i Purdue, blev støttet af LLNL's Laboratory Directed Research and Development Strategic Initiative Program med Lara Leininger, EMC direktør, som hovedefterforsker. Arbejdet involverede at køre storskala all-atom simuleringer på Livermore Computing maskinen Quartz, og disse simuleringer blev udført ved hjælp af computertid givet gennem LLNL's Computational Grand Challenge.
At studere de langvarige afslapningsegenskaber af den kinetiske og potentielle energi i hotspots, holdet udviklede en ny metode kaldet Shock Trapping Internal Boundaries.
"Generelt, stødsimuleringer er begrænset i tid til, når en stødbølge når nedstrøms simuleringsgrænsen, som genererer statsændrende refleksionsbølger, "Sagde Hamilton." I vores metode, vi kan isolere hotspottet, eller en hvilken som helst region af interesse, forhindrer refleksioner i at interagere med det for at tillade kontinuerlig undersøgelse af tidsudvikling. "
Dette gjorde det muligt for teamet at kvantificere hastighederne for afslapning af kinetisk og potentiel energi for at bestemme, at den potentielle energi af hotspottet fortsætter efter at termisk ledning spreder den kinetiske energi.
Molekylær dynamik simuleringer forudsiger, at mere potentiel energi er lokaliseret i hotspots, end deres kinetiske energi (eller temperatur) antyder. Overskydende potentiel energi er bundet til vedvarende anstrengte molekylære tilstande, der er klaret til kemiske reaktioner og forklarer, hvorfor hotspots reagerer hurtigere end hovedparten.
 Varme artikler
Varme artikler
-
 Fluorescerende sonder for at studere cellulær aktivitetFigur viser målretning af cPLA2 af den nydesignede inhibitor og substratsonde. Til venstre:Identificering af forskelle i cPLA2 -niveau i ubehandlet og Trichostatin A (TSA, en inhibitorforbindelse) -be
Fluorescerende sonder for at studere cellulær aktivitetFigur viser målretning af cPLA2 af den nydesignede inhibitor og substratsonde. Til venstre:Identificering af forskelle i cPLA2 -niveau i ubehandlet og Trichostatin A (TSA, en inhibitorforbindelse) -be -
 Multimodal røntgen- og elektronmikroskopi af Allende-meteorittenEksperimentel carbon channel EDS tilt serie. Alle 20 forarbejdede carbon EDS -fremskrivninger bruges som input til GENFIRE -genopbygning. Hvert billede blev maskeret, normaliseret til referenceprojekt
Multimodal røntgen- og elektronmikroskopi af Allende-meteorittenEksperimentel carbon channel EDS tilt serie. Alle 20 forarbejdede carbon EDS -fremskrivninger bruges som input til GENFIRE -genopbygning. Hvert billede blev maskeret, normaliseret til referenceprojekt -
 En enkel måde at få komplekse halvledere til at samle sig selvEt diagram viser, hvordan lag af to 2D-materialer – en perovskit (blå) og et metalhalogenid (gul) – samler sig ud af kemikalier, der vælter rundt i vand (til venstre). Forsamlingen styres af linkermol
En enkel måde at få komplekse halvledere til at samle sig selvEt diagram viser, hvordan lag af to 2D-materialer – en perovskit (blå) og et metalhalogenid (gul) – samler sig ud af kemikalier, der vælter rundt i vand (til venstre). Forsamlingen styres af linkermol -
 Ny metode til asymmetrisk N, N-acetalsyntese lover fremskridt inden for lægemiddeludviklingKredit:Nagoya Institute of Technology Mange af vores lægemidler og andre bioaktive lægemidler er baseret på kemiske strukturer kaldet enantiomerer - molekyler, der er spejlbilleder af hinanden og
Ny metode til asymmetrisk N, N-acetalsyntese lover fremskridt inden for lægemiddeludviklingKredit:Nagoya Institute of Technology Mange af vores lægemidler og andre bioaktive lægemidler er baseret på kemiske strukturer kaldet enantiomerer - molekyler, der er spejlbilleder af hinanden og