


Accelererende geometrioptimering i molekylær simulering
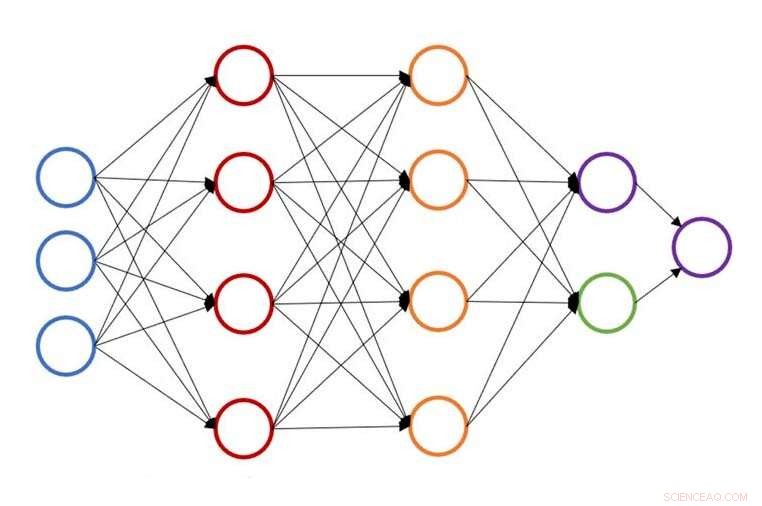
Illustration af en enkelt neural netværksstruktur til forudsigelse af atomenergi. Kredit:John Kitchin, Carnegie Mellon University
Maskinelæring, en dataanalysemetode, der bruges til at automatisere analytisk modelopbygning, har omformet den måde, videnskabsmænd og ingeniører udfører forskning på. En gren af kunstig intelligens (AI) og datalogi, metoden er afhængig af et stort antal algoritmer og brede datasæt til at identificere mønstre og træffe vigtige forskningsbeslutninger.
Anvendelser af maskinlæringsteknikker dukker op inden for overfladekatalyse, muliggør mere omfattende simuleringer af nanopartikler, undersøgelser af segregation, strukturoptimering, on-the-fly indlæring af kraftfelter, og screening med høj gennemstrømning. Imidlertid, at arbejde gennem store mængder data kan ofte være en lang og beregningsmæssigt dyr opgave.
Geometri optimering, ofte det hastighedsbegrænsende trin i molekylære simuleringer, er en vigtig del af beregningsmaterialer og overfladevidenskab. Det giver forskere mulighed for at finde grundtilstands atomare strukturer og reaktionsveje, egenskaber, der bruges til at estimere de kinetiske og termodynamiske egenskaber af molekylære og krystalstrukturer. Mens vital, processen kan være relativt langsom, kræver et stort antal beregninger at gennemføre.
På Carnegie Mellon University, John Kitchin arbejder på at fremskynde denne proces ved at levere en neural netværksbaseret aktiv læringsmetode, der accelererer geometrisk optimering for flere konfigurationer samtidigt. Den nye model sænker antallet af tæthedsfunktionel teori (DFT) eller effektiv medium teori (EMT) beregninger med 50 til 90 procent, giver forskere mulighed for at udføre det samme arbejde på kortere tid eller mere arbejde på samme tid.
"Normalt, når vi laver geometrioptimering, vi starter fra bunden, " sagde Kitchin. "Beregningerne drager sjældent nytte af noget, vi vidste tidligere."
"Ved at tilføje en surrogatmodel til processen, vi har gjort det muligt at stole på tidligere beregninger, i stedet for at starte fra bunden hver gang."
Undersøgelsen illustrerer accelerationen på flere casestudier, inklusive overflader med adsorbater, nøgne metaloverflader, og skubbede elastikbånd til to reaktioner. I hvert tilfælde, Atomic Simulation Environment (ASE)-optimizer Python-pakken tillod færre DFT-beregninger end standardmetoden.
ASE-optimizer Python-pakken er blevet gjort tilgængelig for andre ingeniører og videnskabsmænd for at gøre brugen af neurale netværksensembles aktiv læring til geometrioptimering lettere.
 Varme artikler
Varme artikler
-
 Kemiker forklarer videnskaben bag fyrværkeriUanset om du vil se fyrværkeri langs Bostons Esplanade eller et andet sted den fjerde juli, du vil ikke være den eneste, der slapper af denne ferie. Det er fordi fyrværkeri, forklarede Michael Pollas
Kemiker forklarer videnskaben bag fyrværkeriUanset om du vil se fyrværkeri langs Bostons Esplanade eller et andet sted den fjerde juli, du vil ikke være den eneste, der slapper af denne ferie. Det er fordi fyrværkeri, forklarede Michael Pollas -
 Nobelvindende teknik som Google Earth til molekylerRichard Henderson, en af 2017 nobelprisvindere i kemi, afholder en bakterie-rhodopsin-model forud for en pressekonference på Laboratory of Molecular Biology i Cambridge, England, Onsdag, 4. okt. 201
Nobelvindende teknik som Google Earth til molekylerRichard Henderson, en af 2017 nobelprisvindere i kemi, afholder en bakterie-rhodopsin-model forud for en pressekonference på Laboratory of Molecular Biology i Cambridge, England, Onsdag, 4. okt. 201 -
 Forskere bruger en syntetisk tunge til at sortere whisky fraDette visuelle abstrakt viser et sensorsystem med tre elementer, der kan skelne alder, blandingsstatus, oprindelsesland, og smagselementer i whisky. Kredit:Jinsong Han et al./ Chem 2017 Whisky k
Forskere bruger en syntetisk tunge til at sortere whisky fraDette visuelle abstrakt viser et sensorsystem med tre elementer, der kan skelne alder, blandingsstatus, oprindelsesland, og smagselementer i whisky. Kredit:Jinsong Han et al./ Chem 2017 Whisky k -
 Direkte syntese af hydrogenperoxid ved hjælp af TS-1 understøttede katalysatorerHAADF-billeder af 2,4% Au - 2,4% Pd - 0,2% Pt / TS-1 calcineret prøve. Kredit:Dr Qian He Hydrogenperoxid (H 2 O 2 ) har mange industrielle anvendelser, fra vandbehandling og blegning til kemis
Direkte syntese af hydrogenperoxid ved hjælp af TS-1 understøttede katalysatorerHAADF-billeder af 2,4% Au - 2,4% Pd - 0,2% Pt / TS-1 calcineret prøve. Kredit:Dr Qian He Hydrogenperoxid (H 2 O 2 ) har mange industrielle anvendelser, fra vandbehandling og blegning til kemis
- TikTok:verdens mest værdifulde startup, som du aldrig har hørt om
- Hvilke dyrearter findes der i ferskvandsøkosystemer?
- Forbedret frekvensfordobling tilføjer fotonikværktøjssættet
- Af jordbærgele og jordskælv:Rumstationsundersøgelse studerer kolloider
- Rapporten foreslår skridt til at holde asiatisk karper ude af de store søer
- NASA-NOAA-satellitten ser den nye tropiske storm Rene skylle Cabo Verde-øerne