


En berømt kunstig intelligens har lært et nyt trick:Sådan laver du kemi
At finde ud af, hvad der får nogle proteiner til at lyse, kræver en forståelse af kemi. Kredit:eLife - tidsskriftet, CC BY-SA
Kunstig intelligens har ændret den måde, videnskab udføres på, ved at give forskere mulighed for at analysere de enorme mængder data, moderne videnskabelige instrumenter genererer. Den kan finde en nål i en million høstakke af information, og ved hjælp af dyb læring kan den lære af selve dataene. AI accelererer fremskridt inden for genjagt, medicin, lægemiddeldesign og skabelsen af organiske forbindelser.
Deep learning bruger algoritmer, ofte neurale netværk, der er trænet på store mængder data, til at udtrække information fra nye data. Det er meget forskelligt fra traditionel databehandling med dens trinvise instruktioner. Tværtimod lærer den af data. Dyb læring er langt mindre gennemsigtig end traditionel computerprogrammering og efterlader vigtige spørgsmål – hvad har systemet lært, hvad ved det?
Som kemiprofessor kan jeg godt lide at designe test, der har mindst ét vanskeligt spørgsmål, der strækker de studerendes viden for at fastslå, om de kan kombinere forskellige ideer og syntetisere nye ideer og koncepter. Vi har udtænkt et sådant spørgsmål til plakatbarnet af AI-fortalere, AlphaFold, som har løst problemet med proteinfoldning.
Proteinfoldning
Proteiner er til stede i alle levende organismer. De giver cellerne struktur, katalyserer reaktioner, transporterer små molekyler, fordøjer mad og gør meget mere. De består af lange kæder af aminosyrer som perler på en snor. Men for at et protein kan udføre sit arbejde i cellen, skal det vrides og bøjes til en kompleks tredimensionel struktur, en proces kaldet proteinfoldning. Fejlfoldede proteiner kan føre til sygdom.
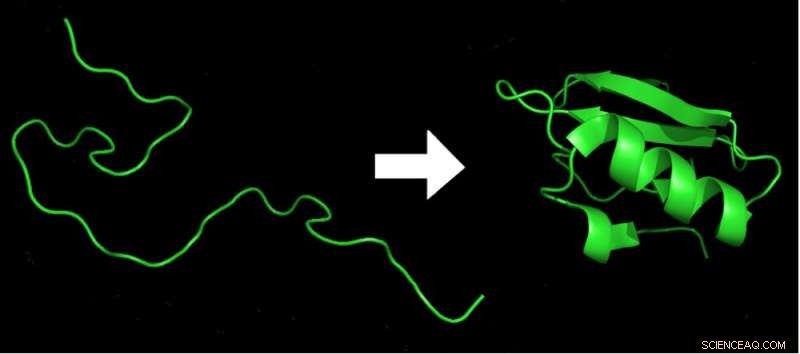
Inden for millisekunder efter udgangen af en aminosyrekæde (venstre) fra ribosomet, foldes den til den laveste energi 3D-form (højre), som er nødvendig for proteinets funktion. Kredit:Marc Zimmer, CC BY-ND
I sin kemi Nobel-modtagelsestale i 1972 postulerede Christiaan Anfinsen, at det burde være muligt at beregne den tredimensionelle struktur af et protein ud fra sekvensen af dets byggesten, aminosyrerne.
Ligesom rækkefølgen og afstanden mellem bogstaverne i denne artikel giver det mening og budskab, så bestemmer rækkefølgen af aminosyrerne proteinets identitet og form, hvilket resulterer i dets funktion.
På grund af den iboende fleksibilitet af aminosyrebyggestenene kan et typisk protein antage 10 til 300 forskellige former. Dette er et enormt antal, mere end antallet af atomer i universet. Men inden for et millisekund vil hvert protein i en organisme foldes til sin helt egen specifikke form - det laveste energiarrangement af alle de kemiske bindinger, der udgør proteinet. Skift kun én aminosyre i de hundredvis af aminosyrer, der typisk findes i et protein, og det kan folde forkert og ikke længere virke.
AlphaFold
I 50 år har dataloger forsøgt at løse problemet med proteinfoldning - med ringe succes. Så i 2016 indledte DeepMind, et AI-datterselskab af Googles moderselskab Alphabet, sit AlphaFold-program. Den brugte proteindatabanken som sit træningssæt, som indeholder de eksperimentelt bestemte strukturer af mere end 150.000 proteiner.
Neuroner, der udtrykker fluorescerende proteiner, afslører hjernestrukturerne af to frugtfluelarver. Kredit:Wen Lu og Vladimir I. Gelfand, Feinberg School of Medicine, Northwestern University
På mindre end fem år havde AlphaFold problemet med proteinfoldning – i det mindste den mest nyttige del af det, nemlig at bestemme proteinstrukturen ud fra dets aminosyresekvens. AlphaFold forklarer ikke, hvordan proteinerne foldes så hurtigt og præcist. Det var en stor sejr for kunstig intelligens, fordi det ikke kun opnåede enorm videnskabelig prestige, det var også et stort videnskabeligt fremskridt, der kunne påvirke alles liv.
I dag, takket være programmer som AlphaFold2 og RoseTTAFold, kan forskere som mig bestemme den tredimensionelle struktur af proteiner ud fra sekvensen af aminosyrer, der udgør proteinet – uden omkostninger – på en time eller to. Før AlphaFold2 skulle vi krystallisere proteinerne og løse strukturerne ved hjælp af røntgenkrystallografi, en proces, der tog måneder og kostede titusindvis af dollars pr. struktur.
Vi har nu også adgang til AlphaFold Protein Structure Database, hvor Deepmind har deponeret 3D-strukturerne af næsten alle de proteiner, der findes i mennesker, mus og mere end 20 andre arter. Til dato har de løst mere end en million strukturer og planlægger at tilføje yderligere 100 millioner strukturer alene i år. Viden om proteiner er steget i vejret. Strukturen af halvdelen af alle kendte proteiner vil sandsynligvis være dokumenteret inden udgangen af 2022, blandt dem mange nye unikke strukturer forbundet med nye nyttige funktioner.
Tænker som en kemiker
AlphaFold2 var ikke designet til at forudsige, hvordan proteiner ville interagere med hinanden, men det har været i stand til at modellere, hvordan individuelle proteiner kombineres til at danne store komplekse enheder sammensat af flere proteiner. Vi havde et udfordrende spørgsmål til AlphaFold – havde dets strukturelle træningssæt lært det noget kemi? Kunne det fortælle, om aminosyrer ville reagere med hinanden - en sjælden, men vigtig begivenhed?
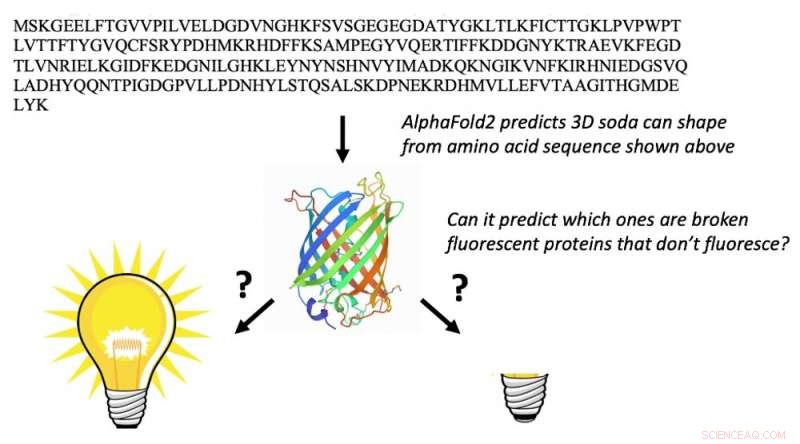
AlphaFold2 kan tage aminosyresekvensen af fluorescerende proteiner (bogstaver øverst) og forudsige deres 3D-tøndeformer (midten). Dette er ikke overraskende. Det helt uventede er, at det også kan forudsige, hvilke fluorescerende proteiner der er 'brudt' og ikke kan fluorescere. Kredit:Marc Zimmer, CC BY-ND
Jeg er en beregningskemiker interesseret i fluorescerende proteiner. Disse er proteiner, der findes i hundredvis af marine organismer som vandmænd og koraller. Deres glød kan bruges til at belyse og studere sygdomme.
Der er 578 fluorescerende proteiner i proteindatabanken, hvoraf 10 er "brudt" og ikke fluorescerer. Proteiner angriber sjældent sig selv, en proces kaldet autokatalytisk posttranslationsmodifikation, og det er meget svært at forudsige, hvilke proteiner der vil reagere med sig selv, og hvilke der ikke vil.
Kun en kemiker med en betydelig mængde fluorescerende proteinkendskab ville være i stand til at bruge aminosyresekvensen til at finde de fluorescerende proteiner, der har den rigtige aminosyresekvens til at gennemgå de kemiske transformationer, der kræves for at gøre dem fluorescerende. Da vi præsenterede AlphaFold2 for sekvenserne af 44 fluorescerende proteiner, der ikke er i proteindatabanken, foldede den de fikserede fluorescerende proteiner anderledes end de ødelagte.
Resultatet overraskede os:AlphaFold2 havde lært noget kemi. Den havde fundet ud af, hvilke aminosyrer i fluorescerende proteiner, der laver den kemi, der får dem til at lyse. Vi har mistanke om, at proteindatabankens træningssæt og flere sekvensjusteringer gør det muligt for AlphaFold2 at "tænke" som kemikere og lede efter de aminosyrer, der kræves for at reagere med hinanden for at gøre proteinet fluorescerende.
Et foldeprogram, der lærer noget kemi fra sit træningssæt, har også bredere implikationer. Ved at stille de rigtige spørgsmål, hvad kan man så ellers få ud af andre deep learning-algoritmer? Kunne ansigtsgenkendelsesalgoritmer finde skjulte markører for sygdomme? Kunne algoritmer designet til at forudsige forbrugsmønstre blandt forbrugere også finde en tilbøjelighed til mindre tyveri eller bedrag? Og vigtigst af alt, er denne evne – og lignende spring i evner i andre AI-systemer – ønskelig? + Udforsk yderligere
Kan proteiner huske det?
Denne artikel er genudgivet fra The Conversation under en Creative Commons-licens. Læs den originale artikel. 
 Varme artikler
Varme artikler
-
 Forskere i verden afslører først strukturen af et vigtigt lægemiddelmålKredit:Monash University Monash University-forskere har brugt en ny teknik til at afsløre strukturen af et vigtigt lægemiddelmål, åbner vejen for forbedrede behandlinger af kroniske sygdomme som
Forskere i verden afslører først strukturen af et vigtigt lægemiddelmålKredit:Monash University Monash University-forskere har brugt en ny teknik til at afsløre strukturen af et vigtigt lægemiddelmål, åbner vejen for forbedrede behandlinger af kroniske sygdomme som -
 Nøgne molekyler, der danser i væske, bliver synligeGrafenlommen. IBS -forskere producerede tynde, men robuste grafenlommer (øverst) for at visualisere bevægelige molekyler under et standardtransmissionselektronmikroskop (TEM). Grafenlagene indeholder
Nøgne molekyler, der danser i væske, bliver synligeGrafenlommen. IBS -forskere producerede tynde, men robuste grafenlommer (øverst) for at visualisere bevægelige molekyler under et standardtransmissionselektronmikroskop (TEM). Grafenlagene indeholder -
 Simuleret kemisk dampaflejring fra en wolframcarbonitridforløberKredit: European Journal of Inorganic Chemistry Tynde film spiller en central rolle i produktionen af elektronik. De kan dyrkes direkte på en substratoverflade gennem processen med kemisk dampa
Simuleret kemisk dampaflejring fra en wolframcarbonitridforløberKredit: European Journal of Inorganic Chemistry Tynde film spiller en central rolle i produktionen af elektronik. De kan dyrkes direkte på en substratoverflade gennem processen med kemisk dampa -
 Spinding af højstyrke polymer nanofibreRamesh Shrestha, Maarten de Boer, og Sheng Shen har transformeret polymerer fra bløde og termisk isolerende materialer til et ultra-stærkt og termisk ledende materiale. Kredit:College of Engineering,
Spinding af højstyrke polymer nanofibreRamesh Shrestha, Maarten de Boer, og Sheng Shen har transformeret polymerer fra bløde og termisk isolerende materialer til et ultra-stærkt og termisk ledende materiale. Kredit:College of Engineering,
- Vil det betale sig for Californien at åbne en kulstoffattig vej?
- Strategi- og struktursammenbrud gavner virksomheder under radikale forandringer
- Undersøgelse kaster mere lys over stjernepopulationer af NGC 6822
- Hvilke molekyler kan passere gennem plasmamembranen uden hjælp?
- Generaliseret Hardys -paradoks viser en endnu stærkere konflikt mellem kvante- og klassisk fysik
- Verdens vinindustri er ved at tilpasse sig klimaforandringerne