


Forudsigelse af den mest stabile bornitridstruktur med kvantesimuleringer
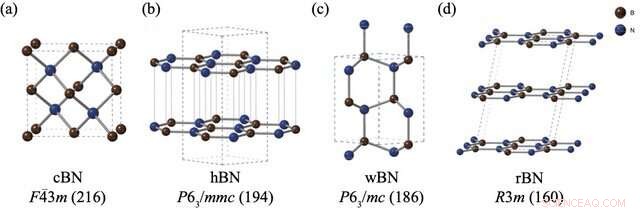
Strukturerne og rumgrupperne af (a) zink-blende bornitrid (cBN), (b) hexagonal bornitrid (hBN), (c) wurtzite bornitrid (wBN) og (d) romboedral bornitrid (rBN). Bor- og nitrogenatomer er afbildet i henholdsvis brunt og blåt. Kredit:Kousuke Nakano fra JAIST.
Bornitrid (BN) er et alsidigt materiale med anvendelser inden for en række ingeniør- og videnskabelige områder. Dette skyldes i høj grad en interessant egenskab ved BN kaldet "polymorfi", karakteriseret ved evnen til at krystallisere til mere end én type struktur. Dette sker generelt som en reaktion på ændringer i temperatur, tryk eller begge dele. Desuden adskiller de forskellige strukturer, kaldet "polymorfer", sig bemærkelsesværdigt i deres fysiske egenskaber på trods af at de har den samme kemiske formel. Som et resultat spiller polymorfer en vigtig rolle i materialedesign, og en viden om, hvordan man selektivt favoriserer dannelsen af den ønskede polymorf, er afgørende i denne henseende.
Imidlertid udgør BN-polymorfer et særligt problem. På trods af udførelse af flere eksperimenter for at vurdere de relative stabiliteter af BN polymorfer, er der ikke opstået en konsensus om dette emne. Mens beregningsmetoder ofte er den foretrukne tilgang til disse problemer, har BN-polymorfer stillet alvorlige udfordringer til standardberegningsteknikker på grund af de svage "van der Waals (vdW) interaktioner" mellem deres lag, hvilket ikke tages højde for i disse beregninger. Desuden manifesterer de fire stabile BN-polymorfer, nemlig rhomboedral (rBN), hexagonal (hBN), wurtzite (wBN) og zink-blende (cBN), inden for et snævert energiområde, hvilket gør indfangning af små energiforskelle sammen med vdW-interaktioner endnu mere udfordrende.
Et internationalt forskerhold ledet af adjunkt Kousuke Nakano fra Japan Advanced Institute of Science and Technology (JAIST) har nu leveret beviser for at afgøre debatten. I deres undersøgelse behandlede de problemet med en state-of-the-art beregningsramme for første principper, nemlig fix-node diffusion Monte Carlo (FNDMC) simuleringer. FNDMC repræsenterer et trin i den populære kvante-Monte Carlo-simuleringsmetode, hvor en parametriseret mange-legeme kvante-"bølgefunktion" først optimeres til at opnå grundtilstanden og derefter leveres til FNDMC.
Derudover beregnede holdet også Gibbs-energien (det nyttige arbejde, der kan opnås fra et system ved konstant tryk og temperatur) af BN-polymorfer for forskellige temperaturer og tryk ved hjælp af tæthedsfunktionel teori (DFT) og fononberegninger. Denne artikel blev gjort tilgængelig online den 24. marts 2022 offentliggjort i The Journal of Physical Chemistry C .
Ifølge FNDMC-resultaterne var hBN den mest stabile struktur efterfulgt af rBN, cBN og wBN. Disse resultater var konsistente ved både 0 K og 300 K (stuetemperatur). DFT-estimationerne gav dog modstridende resultater for to forskellige tilnærmelser. Dr. Nakano forklarer disse modstridende resultater:"Vores resultater afslører, at estimeringen af relative stabiliteter i høj grad påvirkes af den udvekslingskorrelationelle funktionelle eller den tilnærmelse, der bruges i DFT-beregningen. Som et resultat kan en kvantitativ konklusion ikke nås ved hjælp af DFT-fund, og en mere præcis tilgang, såsom FNDMC, er påkrævet."
Navnlig var FNDMC-resultaterne i overensstemmelse med det, der blev genereret af andre raffinerede beregningsmetoder, såsom "koblet klynge", hvilket tyder på, at FNDMC er et effektivt værktøj til at håndtere polymorfer, især dem, der styres af vdW-kræfter. Holdet viste også, at det kan give andre vigtige oplysninger, såsom pålidelige referenceenergier, når eksperimentelle data er utilgængelige.
Dr. Nakano er begejstret for metodens fremtidsudsigter inden for materialevidenskab. "Vores undersøgelse viser FNDMC's evne til at opdage små energiændringer, der involverer vdW-kræfter, hvilket vil stimulere brugen af denne metode til andre van der Waals-materialer," siger han. "Desuden kunne molekylære simuleringer baseret på denne nøjagtige og pålidelige metode styrke materialedesign, hvilket muliggør udvikling af medicin og katalysatorer." + Udforsk yderligere
Forøgelse af nøjagtigheden af atomkraftberegninger med space-warp-koordinattransformation
 Varme artikler
Varme artikler
-
 Nøglen er i belægningen:Flerlagsbelægning for at forbedre korrosionsbestandigheden af stålFlerlagsbelægningen har vist høj modstandsdygtighed over for rust og kan have vidtrækkende konsekvenser på byggeområdet. Kredit:Korea Maritime &Ocean University Stålets styrke gør det til et af de
Nøglen er i belægningen:Flerlagsbelægning for at forbedre korrosionsbestandigheden af stålFlerlagsbelægningen har vist høj modstandsdygtighed over for rust og kan have vidtrækkende konsekvenser på byggeområdet. Kredit:Korea Maritime &Ocean University Stålets styrke gør det til et af de -
 Et skridt nærmere bæredygtig energi fra havvandUnder elektrolyse af vand, elektricitet ledes gennem vandet for at spalte det i andre stoffer. I den ønskede reaktion, flydende vand (H2O) spaltes i oxygengas (O2) og brintgas (H2). I saltvand, natriu
Et skridt nærmere bæredygtig energi fra havvandUnder elektrolyse af vand, elektricitet ledes gennem vandet for at spalte det i andre stoffer. I den ønskede reaktion, flydende vand (H2O) spaltes i oxygengas (O2) og brintgas (H2). I saltvand, natriu -
 Driver fremtiden med revolutionerende lithium -ekstraktionsteknikKredit:CC0 Public Domain Et internationalt forskerhold har været banebrydende og patenteret en ny filtreringsteknik, der en dag kan reducere lithium -ekstraktionstider og ændre den måde, fremtiden
Driver fremtiden med revolutionerende lithium -ekstraktionsteknikKredit:CC0 Public Domain Et internationalt forskerhold har været banebrydende og patenteret en ny filtreringsteknik, der en dag kan reducere lithium -ekstraktionstider og ændre den måde, fremtiden -
 Efter madlavning, bioberigede majs og æg bevarer de nødvendige næringsstoffer for at forhindre bl…Kredit:American Chemical Society Berigede og bioberigede fødevarer er på forkant med indsatsen for at bekæmpe A-vitaminmangel på verdensplan. Men lidt er kendt om, hvilken indflydelse forarbejdnin
Efter madlavning, bioberigede majs og æg bevarer de nødvendige næringsstoffer for at forhindre bl…Kredit:American Chemical Society Berigede og bioberigede fødevarer er på forkant med indsatsen for at bekæmpe A-vitaminmangel på verdensplan. Men lidt er kendt om, hvilken indflydelse forarbejdnin
- Galaktiske vinde får forskere til at undersøge galakser i en hidtil uset skala
- Fakta om og årsager til vulkaner
- Otzi ismanden spiste et fedtfattigt sidste måltid
- Menneskelig adfærd:hvad videnskabsmænd har lært om det fra pandemien
- Radar-lidar-forhold:prospektiv metode til forskning i cirrusskyer
- Hvad er fordelene ved prokaryoter?