


Forskere anvender kvanteberegningsmetoder til forudsigelse af proteinstruktur
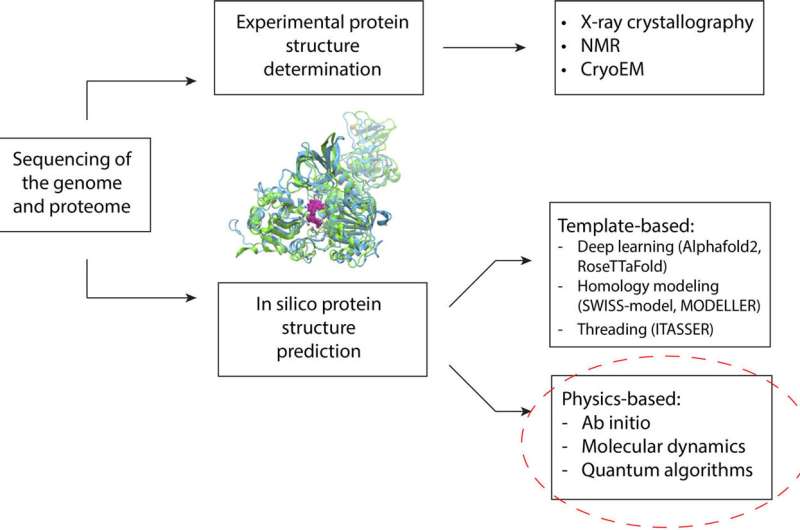
Forskere fra Cleveland Clinic og IBM har for nylig offentliggjort resultater i Journal of Chemical Theory and Computation som kunne danne grundlaget for at anvende kvanteberegningsmetoder til forudsigelse af proteinstruktur.
I årtier har forskere udnyttet beregningsmæssige tilgange til at forudsige proteinstrukturer. Et protein folder sig selv til en struktur, der bestemmer, hvordan det fungerer og binder sig til andre molekyler i kroppen. Disse strukturer bestemmer mange aspekter af menneskers sundhed og sygdom.
Ved præcist at forudsige et proteins struktur kan forskerne bedre forstå, hvordan sygdomme spredes, og dermed hvordan man udvikler effektive terapier. Cleveland Clinic postdoc-stipendiat Bryan Raubenolt, Ph.D. og IBM-forsker Hakan Doga, ph.d. stod i spidsen for et team for at opdage, hvordan kvanteberegning kan forbedre nuværende metoder.
I de senere år har maskinlæringsteknikker gjort betydelige fremskridt i forudsigelse af proteinstruktur. Disse metoder er afhængige af træningsdata (en database med eksperimentelt bestemte proteinstrukturer) til at lave forudsigelser. Det betyder, at de er begrænset af, hvor mange proteiner de er blevet lært at genkende. Dette kan føre til lavere niveauer af nøjagtighed, når programmerne/algoritmerne støder på et protein, der er muteret eller meget forskelligt fra dem, de blev trænet i, hvilket er almindeligt med genetiske lidelser.
Den alternative metode er at simulere fysikken i proteinfoldning. Simuleringer giver forskere mulighed for at se på et givet proteins forskellige mulige former og finde den mest stabile. Den mest stabile form er afgørende for lægemiddeldesign.
Udfordringen er, at disse simuleringer er næsten umulige på en klassisk computer, ud over en vis proteinstørrelse. På en måde kan en forøgelse af størrelsen af målproteinet sammenlignes med at øge dimensionerne af en Rubiks terning. For et lille protein med 100 aminosyrer ville en klassisk computer have brug for den tid, der svarer til universets alder, for at udtømmende søge alle mulige resultater, siger Dr. Raubenolt.
For at hjælpe med at overvinde disse begrænsninger anvendte forskerholdet en blanding af kvante- og klassiske beregningsmetoder. Denne ramme kunne give kvantealgoritmer mulighed for at adressere de områder, der er udfordrende for state-of-the-art klassisk databehandling, herunder proteinstørrelse, iboende lidelse, mutationer og fysikken involveret i proteinfoldning. Rammerne blev valideret ved nøjagtigt at forudsige foldningen af et lille fragment af et Zika-virusprotein på en kvantecomputer sammenlignet med avancerede klassiske metoder.
Den kvante-klassiske hybridrammes indledende resultater overgik både en klassisk fysik-baseret metode og AlphaFold2. Selvom sidstnævnte er designet til at fungere bedst med større proteiner, demonstrerer det ikke desto mindre denne rammes evne til at skabe nøjagtige modeller uden direkte at stole på væsentlige træningsdata.
Forskerne brugte en kvantealgoritme til først at modellere den laveste energikonformation for fragmentets rygrad, hvilket typisk er det mest beregningskrævende trin i beregningen. Klassiske tilgange blev derefter brugt til at konvertere resultaterne opnået fra kvantecomputeren, rekonstruere proteinet med dets sidekæder og udføre endelig forfining af strukturen med klassiske molekylærmekaniske kraftfelter.
Projektet viser en af måderne, hvorpå problemer kan dekonstrueres til dele, med kvanteberegningsmetoder, der adresserer nogle dele, og klassiske beregninger andre, for øget nøjagtighed.
"En af de mest unikke ting ved dette projekt er antallet af involverede discipliner," siger Dr. Raubenolt. "Vores teams ekspertise spænder fra beregningsbiologi og kemi, strukturel biologi, software- og automationsteknik, til eksperimentel atom- og kernefysik, matematik og selvfølgelig kvantecomputere og algoritmedesign. Det krævede viden fra hvert af disse områder for at skabe en beregningsramme, der kan efterligne en af de vigtigste processer for menneskeliv."
Holdets kombination af klassiske og kvanteberegningsmetoder er et væsentligt skridt for at fremme vores forståelse af proteinstrukturer, og hvordan de påvirker vores evne til at behandle og forebygge sygdom. Holdet planlægger at fortsætte med at udvikle og optimere kvantealgoritmer, der kan forudsige strukturen af større og mere sofistikerede proteiner.
"Dette arbejde er et vigtigt skridt fremad i at udforske, hvor kvanteberegningsevner kan vise styrker i forudsigelse af proteinstruktur," siger Dr. Doga. "Vores mål er at designe kvantealgoritmer, der kan finde ud af, hvordan man forudsiger proteinstrukturer så realistisk som muligt."
Flere oplysninger: Hakan Doga et al., A Perspective on Protein Structure Prediction Using Quantum Computers, Journal of Chemical Theory and Computation (2024). DOI:10.1021/acs.jctc.4c00067
Leveret af Cleveland Clinic
 Varme artikler
Varme artikler
-
 Laserskrivning kan muliggøre elektronisk næse til multigassensorAlexander Castonguay (til venstre), kandidatstuderende i laboratoriet for adjunkt Lauren Zarzar, og adjunkt Huanyu Larry Cheng brugte denne laseropsætning til deres tværfaglige samarbejde. Kredit:Kelb
Laserskrivning kan muliggøre elektronisk næse til multigassensorAlexander Castonguay (til venstre), kandidatstuderende i laboratoriet for adjunkt Lauren Zarzar, og adjunkt Huanyu Larry Cheng brugte denne laseropsætning til deres tværfaglige samarbejde. Kredit:Kelb -
 Fyrresaftbaseret plastik:En potentiel gamechanger for fremtidens bæredygtige materialerGrafisk abstrakt. Kredit:DOI:10.1021/acsmacrolett.1c00284 I løbet af de sidste 100 år, plastik og polymerer har ændret den måde, verden fungerer på, fra fly og biler til computere og mobiltelefone
Fyrresaftbaseret plastik:En potentiel gamechanger for fremtidens bæredygtige materialerGrafisk abstrakt. Kredit:DOI:10.1021/acsmacrolett.1c00284 I løbet af de sidste 100 år, plastik og polymerer har ændret den måde, verden fungerer på, fra fly og biler til computere og mobiltelefone -
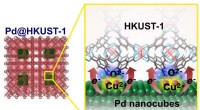 Grænseflade elektronisk tilstand, der forbedrer brintlagringskapacitet i Pd-MOF-materialer(Venstre) Struktur af en Pd@HKUST-1. (Højre) Skematisk diagram, der illustrerer overførslen af elektrisk ladning fra en Pd nanocube til HKUST-1 MOFer (metal-organiske rammer) Kredit:NIMS NIMS, K
Grænseflade elektronisk tilstand, der forbedrer brintlagringskapacitet i Pd-MOF-materialer(Venstre) Struktur af en Pd@HKUST-1. (Højre) Skematisk diagram, der illustrerer overførslen af elektrisk ladning fra en Pd nanocube til HKUST-1 MOFer (metal-organiske rammer) Kredit:NIMS NIMS, K -
 Skinner lys for at lave brintD. desulfuricans-CdS hybrider udviser høj H2 produktionsaktivitet, høj stabilitet og en bemærkelsesværdig effektivitet ved direkte brug af solenergi. Kredit:Inês Cardoso Pereira; Mónica Martins De
Skinner lys for at lave brintD. desulfuricans-CdS hybrider udviser høj H2 produktionsaktivitet, høj stabilitet og en bemærkelsesværdig effektivitet ved direkte brug af solenergi. Kredit:Inês Cardoso Pereira; Mónica Martins De
- Planlægning af orkaner
- Aҫaí bærekstrakter bekæmper malaria hos mus
- Ny undersøgelse belyser design af heterogene katalysatorer til selektiv kuldioxidfotoreduktion
- Cøliakivenlig korn? Forskning viser, at havre kunne være svaret
- Der er nu en tankstation i rummet
- Spansk vulkan i udbrud bliver mere aggressiv:embedsmænd