


En ny ungarsk metode kan hjælpe proteinforskning
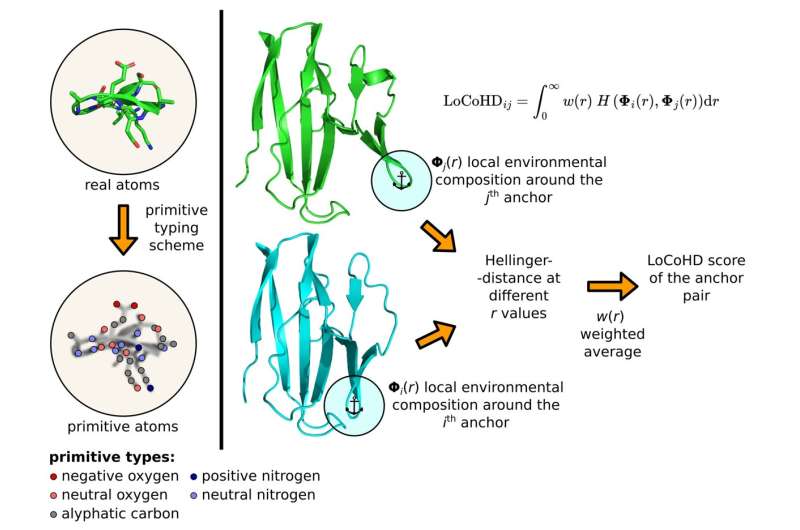
I et papir for nylig offentliggjort i Nature Communications , HUN-REN-ELTE Protein Modeling Research Group (Institute of Chemistry) har lagt grundlaget for en matematisk metode, der muliggør en computerstøttet sammenligning af proteiners tredimensionelle strukturer. Metoden er unik ved, at mens de hidtil tilgængelige alternativer kun har taget højde for atomernes position, omfatter den nye teknik, kaldet LoCoHD (Local Composition Hellinger Distance), også atomernes kemiske information.
Proteiner er molekylære maskiner, der udfører processer, der er nødvendige for, at celler kan fungere, fungerer som molekylære omskiftere, transskriberer information fra DNA, transporterer små og store molekyler og regulerer metabolismerelaterede kemiske reaktioner. Men for at alt dette skal lykkes, skal det pågældende protein have den rette rumlige konformation, dvs. sit eget, korrekte 3D-arrangement.
Adskillige eksperimentelle metoder (røntgenkrystallografi, kernemagnetisk resonansspektroskopi, kryo-elektronmikroskopi) er tilgængelige til at bestemme arrangementet af atomer i et protein, og i løbet af de sidste par årtier har proteinforskere opdaget formen på næsten 220.000 proteiner. Disse resultater kræver i stigende grad udvikling af beregningsmetoder, der er i stand til at analysere disse arrangementer.
En sådan metode er algoritmen kaldet LoCoHD, udviklet af Zsolt Fazekas, en Ph.D. kandidat ved ELTE Hevesy György School of Chemistry og forsker i Dr. András Perczels forskningsgruppe. Algoritmen sammenligner lokale miljøer omkring aminosyrer i proteiner baseret på deres kemiske natur (f.eks. grundstofsammensætning, ladning, hydrofobicitet osv.).
Metoden afgør på en simpel skala fra 0 til 1, hvor forskellige de pågældende strukturer er fra hinanden. Værdier tæt på 0 tyder på en høj lighed mellem atomarrangementer og kemiske egenskaber, mens værdier tæt på 1 indikerer, at de proteiner, der sammenlignes, kan have meget forskellige egenskaber. Den resulterende numeriske værdi (en såkaldt metrik) kan således bruges til at få ny information om det undersøgte system.
Algoritmen bruger en flertrinsprotokol til at generere det tal, der repræsenterer de strukturelle forskelle. I det første trin omdanner den rigtige atomer i proteinet til såkaldte primitive atomer. Disse kan repræsenteres som virtuelt mærkede positioner, hvis mærker fortæller den kemiske natur af det oprindelige atom.
Så for eksempel kan et primitivt atom være et "positivt ladet nitrogen", et "negativt ladet oxygen", et "neutralt ladet oxygen", et "aromatisk kulstof" osv. Mærkerne er genereret efter en såkaldt primitiv typeskema, som fortæller os på en tabuleret måde, hvordan man omdanner rigtige atomer til primitive atomer. Brugeren kan frit specificere denne tabel og fastsætte den kemiske opløsning af metoden.
Det andet trin er at bestemme referencepunkterne for sammenligningen ved at vælge en delmængde af primitive atomer. Disse udvalgte specielle primitive atomer kaldes ankeratomerne. For hvert udvalgt ankeratompar udfører algoritmen et sammenligningstrin, hvis resultat giver det ulighedsmål, vi ønsker. Disse tal kan bruges på lokalt niveau, eller de kan sættes i gennemsnit til en enkelt deskriptor, der karakteriserer hele proteinet.
I undersøgelsen fremhævede forskerne, at metoden også kan bruges i de halvårlige CASP-konkurrencer (Critical Assessment of Protein Structure Prediction), som er en velkendt konkurrence inden for proteinforskning. Under denne begivenhed bruger konkurrenterne forskellige algoritmer til at modellere formen af proteiner med endnu ikke-publicerede strukturer. CASP-dommere bruger en række struktursammenligningsmetoder til at evaluere kandidaterne, men ingen af disse tager højde for kemien i de lokale aminosyremiljøer.
Ved hjælp af data fra 2020 CASP14-konkurrencen har forskerne nu udført sammenlignende analyse af adskillige modellerede proteiner, herunder de strukturer, der er forudsagt af den kunstig intelligens-baserede AlphaFold2-metode. Blandt disse fremhævede de analysen af et protein fra SARS-CoV-2 virus kaldet ORF8. I de modellerede strukturer af dette protein blev aminosyremiljøer identificeret, som adskiller sig væsentligt i deres interaktionsmønstre fra de miljøer, der findes i den eksperimentelle struktur.
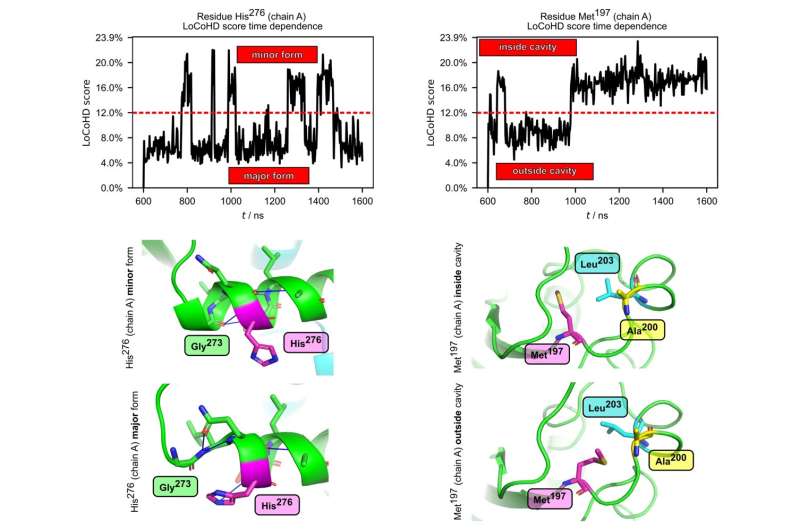
Udover at studere statiske strukturer testede forskerne også, om metoden er egnet til at analysere proteiners indre bevægelse. De brugte simuleringer, der var i stand til at reproducere molekylære bevægelser og data udtrukket fra strukturelle ensembler. Et af de undersøgte systemer var podocin-proteinet, som udfører vitale funktioner i nyren, og hvis mutationer kan forårsage alvorlige, ofte dødelige tilstande.
LoCoHD-metoden blev brugt til at identificere aminosyrer i proteinet, der undergår store kemisk-miljømæssige ændringer under podocins bevægelse, hvilket kan påvirke både dets struktur og funktion. På samme måde er LoCoHD-metoden blevet anvendt med succes i studiet af HIV-1 capsidproteinet, hvor en aminosyre, der er kritisk for dannelsen af viruskappen, er blevet identificeret.
Disse resultater er ikke kun forskningsmæssige kuriositeter, men ved at studere proteinstrukturer mere effektivt kan vi komme tættere på en bedre forståelse af de patogener, der forårsager alvorlige sygdomme, og på at udvikle effektive lægemidler og terapeutika.
Flere oplysninger: Zsolt Fazekas et al., LoCoHD:en metrik til sammenligning af lokale miljøer af proteiner, Nature Communications (2024). DOI:10.1038/s41467-024-48225-0
Journaloplysninger: Nature Communications
Leveret af Eötvös Loránd University
 Varme artikler
Varme artikler
-
 Lavpris, printbar 3-D enhed designet til at analysere kemikalier fra smartphonesKredit: RSC går videre Forskere fra Universitetet i Alicante (Spanien) og Universidad Nacional del Sur (Argentina) har designet og valideret en billig 3D-printet enhed, der tilsluttet en smartpho
Lavpris, printbar 3-D enhed designet til at analysere kemikalier fra smartphonesKredit: RSC går videre Forskere fra Universitetet i Alicante (Spanien) og Universidad Nacional del Sur (Argentina) har designet og valideret en billig 3D-printet enhed, der tilsluttet en smartpho -
 En strandelskers drøm:Et skridt mod langtidsholdbar solcremeKredit:Marina Shemesh/offentligt domæne I en perfekt verden, folk ville flittigt genanvende solskærmen hvert par timer for at beskytte deres sarte hud mod skadelig solstråling. Men i virkeligheden
En strandelskers drøm:Et skridt mod langtidsholdbar solcremeKredit:Marina Shemesh/offentligt domæne I en perfekt verden, folk ville flittigt genanvende solskærmen hvert par timer for at beskytte deres sarte hud mod skadelig solstråling. Men i virkeligheden -
 Forskerhold identificerer en molekylær kode indlejret i protein til regulering af dets glykosylerin…Specifik 29-aminosyresekvens fra LAMP-1 tjener som en Lewis X-kode, som dechifreres af FUT9, og den kan indlejres i erythropoietin for at fremkalde Lewis X-modifikation. Kredit:Nagoya City University
Forskerhold identificerer en molekylær kode indlejret i protein til regulering af dets glykosylerin…Specifik 29-aminosyresekvens fra LAMP-1 tjener som en Lewis X-kode, som dechifreres af FUT9, og den kan indlejres i erythropoietin for at fremkalde Lewis X-modifikation. Kredit:Nagoya City University -
 Kemi giver en ny forsyning af en lovende kræft- og hiv-behandlingKredit:CC0 Public Domain Et lægemiddel isoleret fra en marin skadedyr lover at behandle nogle af verdens mest ubehagelige sygdomme, og forskere ville elske at finde ud af, hvor effektivt det er -
Kemi giver en ny forsyning af en lovende kræft- og hiv-behandlingKredit:CC0 Public Domain Et lægemiddel isoleret fra en marin skadedyr lover at behandle nogle af verdens mest ubehagelige sygdomme, og forskere ville elske at finde ud af, hvor effektivt det er -