


En ny maskinlæringsmodel til karakterisering af materialeoverflader
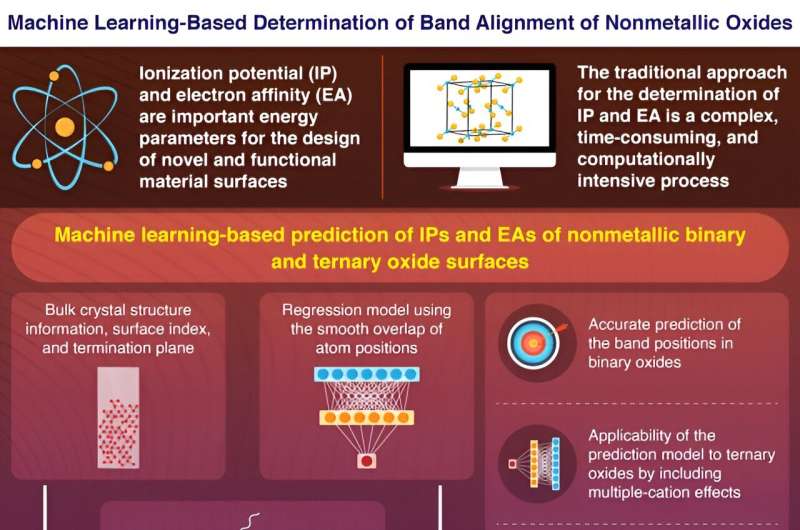
Maskinlæring (ML) muliggør nøjagtig og effektiv beregning af grundlæggende elektroniske egenskaber af binære og ternære oxidoverflader, som vist af forskere fra Tokyo Tech. Deres ML-baserede model kan udvides til andre forbindelser og egenskaber. Resultaterne, offentliggjort i Journal of the American Chemical Society , kunne hjælpe med screening af materialers overfladeegenskaber såvel som i udviklingen af funktionelle materialer.
Design og udvikling af nye materialer med overlegne egenskaber kræver en omfattende analyse af deres atomare og elektroniske strukturer.
Elektronenergiparametre såsom ioniseringspotentiale (IP), den energi, der er nødvendig for at fjerne en elektron fra valensbåndets maksimum, og elektronaffinitet (EA), mængden af energi, der frigives ved binding af en elektron til ledningsbåndets minimum, afslører vigtige information om den elektroniske båndstruktur af overflader af halvledere, isolatorer og dielektriske stoffer.
Den nøjagtige estimering af IP'er og EA'er i sådanne ikke-metalliske materialer kan indikere deres anvendelighed til brug som funktionelle overflader og grænseflader i lysfølsomt udstyr og optoelektroniske enheder.
Derudover afhænger IP'er og EA'er betydeligt af overfladestrukturerne, hvilket tilføjer endnu en dimension til den komplekse procedure for deres kvantificering. Traditionel beregning af IP'er og EA'er involverer brugen af nøjagtige beregninger med første principper, hvor bulk- og overfladesystemerne kvantificeres separat. Denne tidskrævende proces forhindrer kvantificering af IP'er og EA'er for mange overflader, hvilket nødvendiggør brugen af beregningseffektive tilgange.
For at løse de vidtrækkende problemer, der påvirker kvantificeringen af IP'er og EA'er af ikke-metalliske faste stoffer, har et team af forskere fra Tokyo Institute of Technology (Tokyo Tech), ledet af professor Fumiyasu Oba, vendt deres fokus mod ML.
Prof. Oba siger:"I de senere år har ML fået meget opmærksomhed inden for materialevidenskabelig forskning. Evnen til virtuelt at screene materialer baseret på ML-teknologi er en meget effektiv måde at udforske nye materialer med overlegne egenskaber. Også evnen til at træne store datasæt ved hjælp af nøjagtige teoretiske beregninger giver mulighed for vellykket forudsigelse af vigtige overfladekarakteristika og deres funktionelle implikationer."
Forskerne brugte et kunstigt neuralt netværk til at udvikle en regressionsmodel, der inkorporerede den glatte overlapning af atompositioner (SOAP'er) som numeriske inputdata. Deres model forudsagde nøjagtigt og effektivt IP'erne og EA'erne for binære oxidoverflader ved at bruge oplysningerne om bulkkrystalstrukturer og overfladetermineringsplaner.
Desuden kunne den ML-baserede forudsigelsesmodel "overføre læring", et scenarie, hvor en model udviklet til et bestemt formål kan fås til at inkorporere nyere datasæt og genanvendes til yderligere opgaver. Forskerne inkluderede virkningerne af flere kationer i deres model ved at udvikle "lærelige" SOAP'er og forudsagde IP'er og EA'er for ternære oxider ved hjælp af transfer learning.
Prof. Oba konkluderer:"Vores model er ikke begrænset til forudsigelse af overfladeegenskaber af oxider, men kan udvides til at studere andre forbindelser og deres egenskaber."
Flere oplysninger: Shin Kiyohara et al., Band Alignment of Oxides by Learnable Structural-Descriptor-Aided Neural Network and Transfer Learning, Journal of the American Chemical Society (2024). DOI:10.1021/jacs.3c13574
Journaloplysninger: Tidsskrift for American Chemical Society
Leveret af Tokyo Institute of Technology
 Varme artikler
Varme artikler
-
 Teknik giver detaljeret overblik over, hvordan visse polymerer dannes, låse op for svar om kernedan…Forskningen pryder forsiden af Physical Chemistry Chemical Physics. Kredit:Royal Society of Chemistry Forestil dig en lille dråbe. Den indeholder vand, det brusende antiseptiske hydrogenperoxid,
Teknik giver detaljeret overblik over, hvordan visse polymerer dannes, låse op for svar om kernedan…Forskningen pryder forsiden af Physical Chemistry Chemical Physics. Kredit:Royal Society of Chemistry Forestil dig en lille dråbe. Den indeholder vand, det brusende antiseptiske hydrogenperoxid, -
 Forskningsteam afdækker ekstraordinære egenskaber ved strontiumniobatForskere ledet af professor T Venky Venkatesan (første række, centrum), Direktør for NUSNNI, afdækkede ekstraordinære egenskaber ved halvledermateriale strontiumniobat. Kredit:National University of S
Forskningsteam afdækker ekstraordinære egenskaber ved strontiumniobatForskere ledet af professor T Venky Venkatesan (første række, centrum), Direktør for NUSNNI, afdækkede ekstraordinære egenskaber ved halvledermateriale strontiumniobat. Kredit:National University of S -
 Dobbelt glæde:Ny syntetisk transmembran ionkanal kan aktiveres på to måderEn multiblok amfifil VF blev udviklet. Når VF blev inkorporeret i lipid-dobbeltlagsmembranerne, VF dannede en supramolekylær ionkanal. VFs iontransportegenskab kunne ændres reversibelt ved tilsætning
Dobbelt glæde:Ny syntetisk transmembran ionkanal kan aktiveres på to måderEn multiblok amfifil VF blev udviklet. Når VF blev inkorporeret i lipid-dobbeltlagsmembranerne, VF dannede en supramolekylær ionkanal. VFs iontransportegenskab kunne ændres reversibelt ved tilsætning -
 Team skaber mikrober for at omdanne affald til nyttige kemikalierForskere gensplejsede bakterier til produktion af itaconsyre, skabe dynamiske kontroller, der adskiller mikrobielle vækst- og produktionsfaser for øget effektivitet og syreudbytte. Kredit:NREL Et
Team skaber mikrober for at omdanne affald til nyttige kemikalierForskere gensplejsede bakterier til produktion af itaconsyre, skabe dynamiske kontroller, der adskiller mikrobielle vækst- og produktionsfaser for øget effektivitet og syreudbytte. Kredit:NREL Et
- Robotkontrolsystem til at gribe og slippe genstande under både tørre og våde forhold
- Hvordan man estimerer Crowd Density
- Rotation på en otteformet sti
- Ny spektroskopimetode kan føre til bedre optiske enheder
- Forskere demonstrerer farveudskrivning uden blæk med nanomaterialer
- Kunsten at orme gennem trange rum