


Fremskynder opdagelsen af enkeltmolekylemagneter med dyb læring
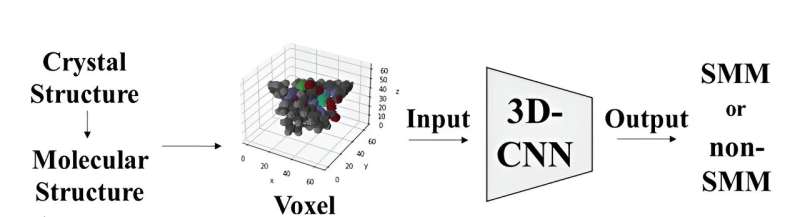
Syntetisering eller undersøgelse af bestemte materialer i laboratoriemiljøer udgør ofte udfordringer på grund af sikkerhedsproblemer, upraktiske eksperimentelle forhold eller omkostningsbegrænsninger. Som reaktion herpå henvender forskerne sig i stigende grad til deep learning-metoder, der involverer udvikling og træning af maskinlæringsmodeller til at genkende mønstre og sammenhænge i data, der omfatter information om materialeegenskaber, sammensætninger og adfærd.
Ved at bruge dyb læring kan videnskabsmænd hurtigt komme med forudsigelser om materialeegenskaber baseret på materialets sammensætning, struktur og andre relevante egenskaber, identificere potentielle kandidater til yderligere undersøgelser og optimere syntesebetingelser.
Nu, i en undersøgelse, der vises i International Union of Crystallography Journal (IUCrJ) , professor Takashiro Akitsu, adjunkt Daisuke Nakane og hr. Yuji Takiguchi fra Tokyo University of Science (TUS) har brugt dyb læring til at forudsige enkeltmolekylemagneter (SMM'er) fra en pulje af 20.000 metalkomplekser. Denne innovative strategi strømliner materialeopdagelsesprocessen ved at minimere behovet for lange eksperimenter.
Enkeltmolekylemagneter (SMM'er) er metalkomplekser, der demonstrerer magnetisk afslapningsadfærd på det individuelle molekyleniveau, hvor magnetiske momenter undergår ændringer eller afslapning over tid. Disse materialer har potentielle anvendelser i udviklingen af højdensitetshukommelse, kvantemolekylære spintroniske enheder og kvantecomputere. SMM'er er karakteriseret ved at have en høj effektiv energibarriere (Ueff ) for at det magnetiske moment kan vende. Disse værdier er dog typisk i intervallet fra ti til hundredvis af Kelvin, hvilket gør SMM'er udfordrende at syntetisere.
Forskerne brugte dyb læring til at identificere forholdet mellem molekylære strukturer og SMM-adfærd i metalkomplekser med salen-ligander. Disse metalkomplekser blev valgt, da de let kan syntetiseres ved at kompleksbinde aldehyder og aminer med forskellige 3d- og 4f-metaller.
For datasættet arbejdede forskerne meget med at screene 800 artikler fra 2011 til 2021, indsamle information om krystalstrukturen og bestemme, om disse komplekser udviste SMM-adfærd. Derudover opnåede de 3D strukturelle detaljer om molekylerne fra Cambridge Structural Database.
Den molekylære struktur af komplekserne blev repræsenteret ved hjælp af voxels eller 3D-pixel, hvor hvert element blev tildelt en unik RGB-værdi. Efterfølgende tjente disse voxel-repræsentationer som input til en 3D Convolutional Neural Network-model baseret på ResNet-arkitekturen. Denne model er specielt designet til at klassificere molekyler som enten SMM'er eller ikke-SMM'er ved at analysere deres 3D-molekylære billeder.
Da modellen blev trænet på et datasæt af krystalstrukturer af metalkomplekser indeholdende komplekser af salen-typen, opnåede den en nøjagtighedsgrad på 70 % ved at skelne mellem de to kategorier. Da modellen blev testet på 20.000 krystalstrukturer af metalkomplekser indeholdende Schiff-baser, opdagede den med succes metalkomplekserne rapporteret som enkeltmolekylemagneter.
"Dette er den første rapport om dyb læring om de molekylære strukturer af SMM'er," siger prof. Akitsu.
Mange af de forudsagte SMM-strukturer involverede multinukleære dysprosiumkomplekser, kendt for deres høje Ueff værdier. Selvom denne metode forenkler SMM-opdagelsesprocessen, er det vigtigt at bemærke, at modellens forudsigelser udelukkende er baseret på træningsdata og ikke eksplicit forbinder kemiske strukturer med deres kvantekemiske beregninger, en foretrukken metode i AI-assisteret molekylært design. Yderligere eksperimentel forskning er påkrævet for at opnå data om SMM-adfærd under ensartede forhold.
Denne forenklede tilgang har dog sine fordele. Det reducerer behovet for komplekse beregninger og undgår den udfordrende opgave at simulere magnetisme.
Prof. Akitsu konkluderer:"At vedtage en sådan tilgang kan vejlede designet af innovative molekyler, hvilket medfører betydelige besparelser i tid, ressourcer og omkostninger i udviklingen af funktionelle materialer."
Flere oplysninger: Yuji Takiguchi et al., Forudsigelsen af enkelt-molekyle magnetegenskaber via dyb læring, IUCrJ (2024). DOI:10.1107/S2052252524000770
Leveret af Tokyo University of Science
 Varme artikler
Varme artikler
-
 Låse dørene op til effektive COVID-19-behandlingerKredit:Majid D. Farahani et al., DOI:10.1002/cmdc.202200092 Et hold af tværfaglige forskere fra Institut National de la Recherche Scientifique (INRS) håber at kunne identificere effektive COVID-19-
Låse dørene op til effektive COVID-19-behandlingerKredit:Majid D. Farahani et al., DOI:10.1002/cmdc.202200092 Et hold af tværfaglige forskere fra Institut National de la Recherche Scientifique (INRS) håber at kunne identificere effektive COVID-19- -
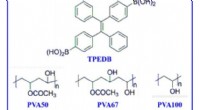 Storskala fremstilling af polymerbaseret stuetemperatur phosphorescens via klikkemiMolekylformler for fosfor og polymermatricer. De molekylære formler for TPEDB, PVA med forskellig alkohollysegrad (PVA50, PVA67, PVA100), og kontrollerede polymerer (PDDA, PSS, og PVDF). Kredit:Scienc
Storskala fremstilling af polymerbaseret stuetemperatur phosphorescens via klikkemiMolekylformler for fosfor og polymermatricer. De molekylære formler for TPEDB, PVA med forskellig alkohollysegrad (PVA50, PVA67, PVA100), og kontrollerede polymerer (PDDA, PSS, og PVDF). Kredit:Scienc -
 3D-ordnet kanal forbedrer elektrokatalyseGrafisk abstrakt. Kredit:DOI:10.1021/jacs.1c04653 Et team ledet af prof. YU Shuhong og prof. HOU Zhonghuai fra University of Science and Technology of China (USTC) fra det kinesiske videnskabsakad
3D-ordnet kanal forbedrer elektrokatalyseGrafisk abstrakt. Kredit:DOI:10.1021/jacs.1c04653 Et team ledet af prof. YU Shuhong og prof. HOU Zhonghuai fra University of Science and Technology of China (USTC) fra det kinesiske videnskabsakad -
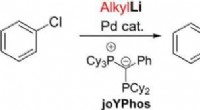 Direkte kobling af arylhalogenider og alkyllithiumforbindelser ved palladiumkatalyseKredit:Wiley Palladiumkatalysatorer hjælper med at syntetisere nøglekemikalier til mange industrier. Imidlertid, direkte reaktion af to grundlæggende reagenser, arylhalogenider og alkyllithiumforb
Direkte kobling af arylhalogenider og alkyllithiumforbindelser ved palladiumkatalyseKredit:Wiley Palladiumkatalysatorer hjælper med at syntetisere nøglekemikalier til mange industrier. Imidlertid, direkte reaktion af to grundlæggende reagenser, arylhalogenider og alkyllithiumforb
- Udvikling af nyt protein kan føre til nye behandlingsmuligheder for kræft, fødselsdefekt
- Mørk energiundersøgelse giver et nyt syn på glorie i mørkt stof, rapporterer fysikere
- Ny elektronmikroskopmetode detekterer magnetisme i atomskala
- Fossil Fakta for Børn
- Undersøgelse bekræfter hårnålehvirvler i supersonisk turbulens
- Human Skull Growth