


Kromosomorganisation kommer ud af 1-D-mønstre
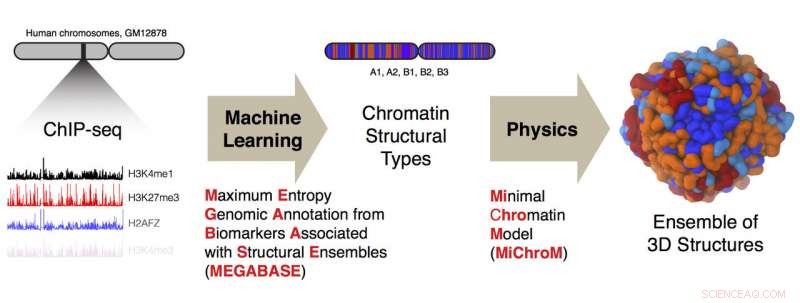
Forskere ved Rice University og Baylor College of Medicine har udviklet en beregningspipeline til at konvertere endimensionelle ChIP-sekventeringsdata om DNA til tredimensionelle strukturer af menneskelige kromosomer. Kredit:Ryan Cheng/Michele Di Pierro
DNA'et i en menneskelig celle er 2 yards (1,83 meter) langt og omslutter millioner af perlelignende histonproteiner for at passe ind i cellens kerne. Forskere ved Rice University og Baylor College of Medicine viste, at undersøgelse af den kemiske tilstand af disse proteiner gør det muligt at forudsige, hvordan et helt DNA-kromosom vil foldes.
Forskere baseret på Rice's Center for Theoretical Biological Physics (CTBP) har konstrueret computermodeller til at analysere epigenetiske mærker, som omfatter proteiner bundet til DNA samt kemiske modifikationer til histonproteiner. De høstede informationen kodet i disse markeringer for at forudsige, hvordan kromosomerne folder sig i tre dimensioner.
Deres resultater flytter genetikområdet tættere på evnen til at forudsige den foldede struktur af hele genomer, som en dag kan hjælpe med at identificere fejlfoldningsrelaterede genetiske sygdomme.
Værket vises i denne uge i Procedurer fra National Academy of Sciences .
Pakket ind i kernen, DNA foldes til en funktionel form, der adskiller sig i forskellige typer celler. Fordi hver celle i en organisme indeholder det samme DNA, epigenetiske mærker hjælper den med at finde den rigtige form til den type celle, den bebor.
"Noget oven på den genetiske kode fortæller cellen, hvad den skal være og bestemmer, hvilke dele af kromosomet der skal læses til enhver tid, "sagde biofysiker Peter Wolynes, medforfatter til papiret. "Det er de såkaldte epigenetiske mærker."
I fællesskab epigenetiske mærker hjælper med at pakke genomet ind i de løse, men højt organiserede rum, det indtager under interfase, den arbejdende "middelalder" i en celles liv. Disse rum bringer transkriptionsrelaterede gener i umiddelbar nærhed og giver dem mulighed for at kommunikere og fungere.
Epigenetiske mærker kan afsløres ved en etableret teknik kaldet ChIP-sekventering, som kortlægger proteinbindingssteder langs DNA.
"Vi forstår ikke præcis, hvordan genomet bliver markeret, men vi kan måle det gennem ChIP-sekventering, som er blevet et ret ligetil eksperiment, " sagde Wolynes. "På samme måde som vi kan se genetisk kode (DNA'et), vi kan også måle disse mærker direkte i mange forskellige celler. De er blevet det næste lag af sekvens på genomet."
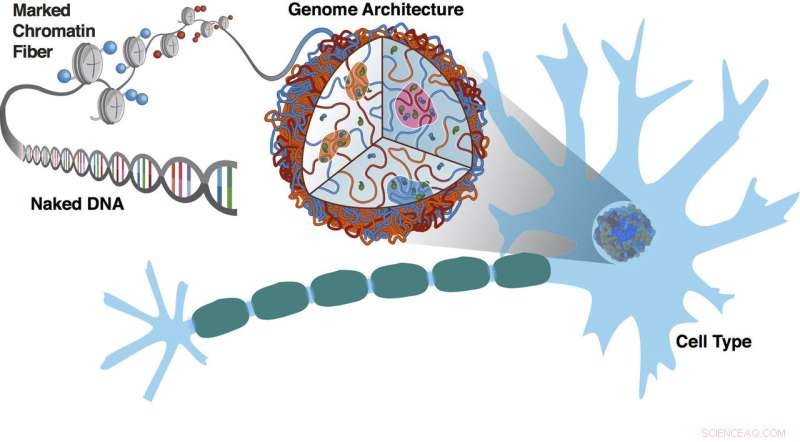
Eksperimenter på Rice University og Baylor College of Medicine viser, hvordan segmenter af kromatin med de samme epigenetiske markeringsmønstre lokaliseres sammen i en proces relateret til faseadskillelse. Nøgent DNA er dekoreret med epigenetiske markeringer, der koder for det tredimensionelle arrangement af kromosomer. Genomarkitekturen og markeringsmønstrene er karakteristika for celletypen, i dette tilfælde en nervecelle med sin karakteristiske myelinskede. Kredit:Sigrid Knemeyer/Center for Teoretisk Biologisk Fysik ved Rice University
"Det er et andet niveau af information, " sagde medforfatter og biofysiker José Onuchic. "Hver en af dine cellers DNA er den samme. Imidlertid, forskellige slags celler har forskellig epigenetik, så deres udtryksmønstre er forskellige."
Co-lead forfattere og Rice postdoc-stipendiater Michele Di Pierro og Ryan Cheng brugte ChIP-sekventeringsdata til en human lymfoblastcelle, der sonderer 84 forskellige DNA-bindende proteiner og 11 kemiske modifikationer af histoner. Histonproteiner hjælper med at organisere genomet ved at fungere som spoler, som DNA vikler sig om.
Ved at bruge data fra kun nogle af kromosomerne, de trænede et brugerdefineret neuralt netværk kaldet MEGABASE (Maximum Entropy Genomic Annotation from Biomarkers Associated with Structural Ensembles) til at udsende en sekvens af kromatintyper. Det afslørede, hvordan de epigenetiske mærker var relateret til kamrene, de sagde. Når først du er blevet trænet, de validerede MEGABASE-modellen ved at tilføre den data fra de resterende kromosomer. Det frembragte et nyt sæt strukturelle typer til analyse af Rice -teamets MiChroM -program, en fætter til laboratoriets AWSEM energilandskabsalgoritme, der forudsiger strukturerne af proteiner. MiChroM-algoritmen forudsagde 3-D-strukturerne af kromosomerne.
"Vores resultater understøtter ideen om, at kompartmentalisering i kromosomer opstår fra faseadskillelse af forskellige kromatintyper i kernen, som adskillelsen af olie og vand, " sagde Cheng.
Da forskerne reducerede det originale datasæt til kun de 11 histonmarkeringer og kørte beregningerne igen, resultaterne var kun marginalt anderledes. Ultimativt, de bestemte histondata alene er tilstrækkelige til at forudsige et kromosoms form. "Der er en veldefineret kode, der forbinder histonmarkeringerne med strukturen, " sagde Di Pierro. "Det er velbevaret, så det er sandsynligt, at det har en funktion."
For at validere deres teori, forskerne sammenlignede deres resultater med kontaktkort over lymfoblastceller genereret af Hi-C. Denne eksperimentelle teknik, som bruger high-throughput sekventering til at identificere foldemønstre i DNA, blev udviklet af medforfatter Erez Lieberman Aiden, direktør for Baylor's Center for Genome Architecture og seniorforsker ved CTBP.
"Dette papir siger, at vi kan tage en-dimensionel information om histoner og bruge den med vores big-data-værktøjer til at forudsige tredimensionel struktur, " sagde Wolynes.
Deres succes får teamet tættere på det endelige mål med en teori, der forudsiger arkitekturen for et helt genom. Imidlertid, et hønen-eller-ægget-problem er tilbage:Folder kromatin sig på grund af markørerne, eller kommer markørerne frem på grund af foldningen?
"Det er alt sammen en del af vores fascination af, hvordan livet fungerer, " sagde Di Pierro. "Det er et smukt problem."
 Varme artikler
Varme artikler
-
 Gennembrud kan hjælpe gartnere med at opnå succes med frøsåningKredit:Royal Holloway, University of London Forskere fra Royal Holloway, University of London, og universitetet i Osnabrück i Tyskland har fundet ud af, at almindelige svampe kunne holde nøglen ti
Gennembrud kan hjælpe gartnere med at opnå succes med frøsåningKredit:Royal Holloway, University of London Forskere fra Royal Holloway, University of London, og universitetet i Osnabrück i Tyskland har fundet ud af, at almindelige svampe kunne holde nøglen ti -
 Eksplosion af rotter, kløver, sengelus, myg utilsigtet evolutionær konsekvens af urbaniseringKredit:CC0 Public Domain Den seneste opstandelse om sæder på en British Airways-flyvning, der kravler med væggelus, er kun en af de utilsigtede konsekvenser, som urbanisering verden over har på
Eksplosion af rotter, kløver, sengelus, myg utilsigtet evolutionær konsekvens af urbaniseringKredit:CC0 Public Domain Den seneste opstandelse om sæder på en British Airways-flyvning, der kravler med væggelus, er kun en af de utilsigtede konsekvenser, som urbanisering verden over har på -
 Marine hvirvelløse dyr har støjende menneskelige naboerStående bølgerør design. Kredit:J.S. Krumholtz, D.M. Hudson, D.L. Pochtar, N.C. Dickenson, G.A. Dossot, E.B. Bager, T.E. Moll Ligesom mennesker, livet i havet oplever konstant stress. De står over
Marine hvirvelløse dyr har støjende menneskelige naboerStående bølgerør design. Kredit:J.S. Krumholtz, D.M. Hudson, D.L. Pochtar, N.C. Dickenson, G.A. Dossot, E.B. Bager, T.E. Moll Ligesom mennesker, livet i havet oplever konstant stress. De står over -
 Hvilke celler kan ses af det menneskelige øje?Levende organismer består af millioner, milliarder eller billioner af celler. Menneskelige kroppe har op til 37 billioner, hvoraf de fleste kun kan ses under et mikroskop. Andre organismer har imid
Hvilke celler kan ses af det menneskelige øje?Levende organismer består af millioner, milliarder eller billioner af celler. Menneskelige kroppe har op til 37 billioner, hvoraf de fleste kun kan ses under et mikroskop. Andre organismer har imid
- Taktisk responsiv Launch-2 nyttelast lanceret i kredsløb efter at være blevet bygget på rekordtid
- Ny tornado-ulykkesanalyse vil forbedre fremtidige forudsigelser
- Lavtlønsarbejdere i fare for automatisering:undersøgelse
- Nye fund om fortiden og fremtiden for havisen i Arktis
- Forskere får fotoner til at interagere, tage et skridt i retning af langlevende kvantehukommelse
- Sådan beregnes procentdel Daglig værdi