


En bedre måde at finde RNA-virusnåle i databasehøstakke
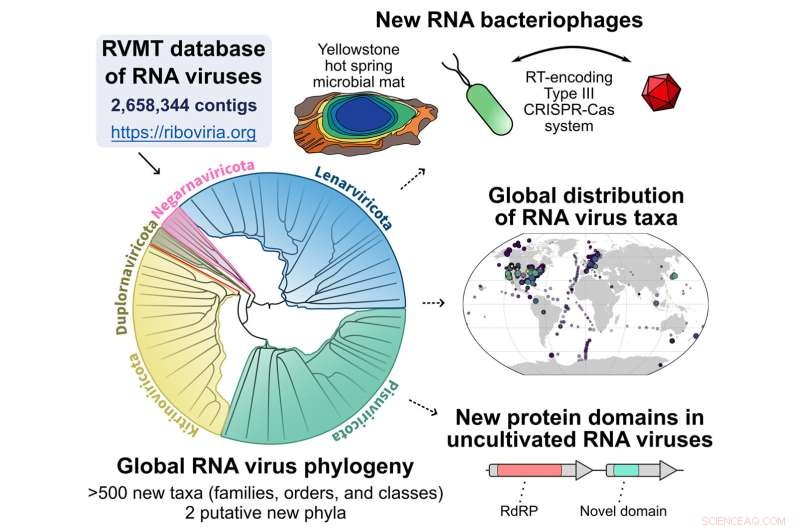
Grafisk oversigt over pipelinen, der starter med RNA Virus MetaTranscriptomes (RVMT) databasen for at afdække udvidelsen i RNA virus diversitet. Kredit:Simon Roux
En zoologisk have tilbød engang en malebog med isbjørne i vinterscener, der kom med farveblyanter i forskellige nuancer af hvid. For forskere, der søger efter sekvenser af RNA-vira i store datasæt, kan deres arbejde svare til at finde et enkelt snefnug på en farvet side i den bog.
Udgivet online 28. september 2022 i Cell , et hold ledet af forskere ved Tel Aviv University i Israel, National Center for Biotechnology Information og U.S. Department of Energy (DOE) Joint Genome Institute (JGI), en DOE Office of Science User Facility beliggende ved Lawrence Berkeley National Laboratory ( Berkeley Lab) beskriver en beregningspipeline, der specifikt kan scanne for disse snefnug eller RNA-virussekvenser. Ved at bruge denne arbejdsgang finkæmmede holdet mere end 5.000 datasæt af RNA-sekvenser (metatranskriptomer) genereret fra forskellige miljøprøver rundt om i verden, hvilket resulterede i en femdobling af RNA-virusdiversiteten.
"Verdenen af vira omkring os er enorm, og vi har nu midlerne til at udforske den," sagde Eugene Koonin, en seniorforsker ved NCBI og en af seniorforfatterne på papiret, om den afdækkede virale mangfoldighed. "Selvom de tekniske udfordringer ved dataanalyse i denne skala er formidable."
Beregningssigter til at filtrere sekvenser
Der er flere mikrober på planeten end partikler i en håndfuld snavs, og vira er langt flere end mikroberne. Fremskridt inden for sekventeringsteknologier og beregningsværktøjer har afsløret en mangfoldighed af vira, der inficerer ikke kun afgrøder, dyr og mennesker, men også mikrober, hvis tilstedeværelse eller fravær kan påvirke planetens næringsstofkredsløb.
Mens de fleste organismers genetiske information er kodet i DNA, hvor RNA leverer instruktionerne inde i DNA til cellen, lagrer RNA-vira deres genetiske information i RNA uden et DNA-stadium. "Jeg vil hævde, at RNA-vira globalt er endnu mindre kendte end DNA-vira," sagde Simon Roux, en JGI-forsker og en af projektets medledere. "Men på samme måde som DNA-vira inficerer RNA-vira mikrober over hele verden og fører til celledød og/eller dybtgående ændringer i cellefysiologien under infektion."
Mens alle RNA-vira har et gen, der koder for et enzym kaldet RNS-dirigeret RNA-polymerase (RdRP), der er nødvendigt for at replikere RNA-genomreplikationen, har det været en udfordring at opdage det. At finde RNA-virussnefnug i snestormen af genomiske data involverede udvikling af specielle beregningssigter til at frafiltrere sekvenser, der sandsynligvis ikke ville indeholde RdRP-sekvensen.
Arbejdet var resultatet af et tre-vejs samarbejde, der begyndte i 2019, mindede om Uri Neri fra Tel Aviv University, en af projektlederne og førsteforfatter til undersøgelsen. Medlemmer af Tel Aviv- og NCBI-holdene, som allerede arbejdede på at udvinde prokaryote vira sammen, lærte af JGI's Nikos Kyrpides, at hans Microbiome Data Science-gruppe også arbejdede på RNA-virusudvinding. Efter et par virtuelle møder mellem de tre teams stod det klart, at en større samarbejdsindsats ville være langt mere effektiv til at opnå resultater af højere kvalitet sammenlignet med mindre individuelle indsatser. Dette er også den type synergistisk og kollaborativ fællesskabsånd, som JGI går ind for og aktivt fremmer.
Holdet brugte alle de offentligt tilgængelige metatranskriptomdatasæt fra JGI's Integrated Microbial Genomes &Microbiomes (IMG/M) system. "Derefter undersøgte vi mange flere prøver og forfinede vores metode," sagde Neri. "Vores team voksede, og det samme gjorde omfanget af projektet." Til dette formål, understregede Kyrpides, kan bidragene fra de talrige JGI-videnskabsbrugere til at indsamle og indsende deres mikrobiomprøver til sekventering på JGI ikke overvurderes. Deres samarbejde og støtte, sagde han, og i flere tilfælde, deres tilladelse til at bruge endnu ikke-publicerede sekvensdata, var helt afgørende for succesen med denne indsats, og det samme var anerkendelsen af deres bidrag.
Både Roux og Koonin bemærkede, at overfloden af afdækkede RNA-virussekvenser "betydeligt ændrer det globale syn på virusdiversitet", dog ikke på det højere niveau af klassifikationer af virusgrupper (phyla.) De nye sekvenser udfylder nogle huller på eksisterende virus grupper, samtidig med at der tilføjes nye filialer. Derudover ser RNA-vira ikke ud til at være jævnt fordelt over hele verden.
En udvidet gruppe er vira forbundet med bakterier; indtil nu har de fleste af de kendte RNA-vira været forbundet med eukaryoter. Sammen med udvidelsen af bakterie-associerede RNA-vira er opdagelsen af, at "nogle få bakterier bruger CRISPR til at forsvare sig mod RNA," bemærkede Roux, "selvom det er uklart, hvorfor dette så sjældent opdages."
Udvikling af tilgange til at forene "rigtige" Big Data
For holdet er det beregningsmæssige arbejde, der førte til den afslørede overflod af RNA-vira, kun begyndelsen. "Jeg siger ofte, at bare det at identificere en sekvens som viral ikke engang er halvdelen af historien." sagde Neri. "Vi investerede en stor del af vores indsats i analyserne efter opdagelsen - så godt vi kunne, forsøgte vi at beskrive de proteindomæner, hver virus bærer, og hvem der er deres sandsynlige vært. Vi har gjort al den information helt fri og åbent. tilgængelig for det bredere videnskabelige samfund."
Uri Gophna fra Tel Aviv University og Koonin bemærkede begge, at anden forskning parallelt har rapporteret lignende "dramatiske udvidelser" af det globale RNA-virom. "Vi er nu nødt til at sammenligne og forene resultaterne og komme frem til et enkelt, ikke-redundant datasæt," sagde Koonin. "Forhåbentlig vil vi relativt snart være i stand til at estimere den faktiske størrelse af RNA-viromet. Men dette er nu ægte Big Data, vi har at gøre med milliarder af sekvenser og snart med billioner. Udviklingen af effektive, automatiserede tilgange til analyse og klassificering af sekvensdata i denne skala er afgørende." + Udforsk yderligere
Et automatiseret værktøj til vurdering af virusdatakvalitet
Sidste artikelDe mystiske Denisovans
Næste artikelNy cellefri proteinkrystalliseringsmetode til fremme af strukturbiologi
 Varme artikler
Varme artikler
-
 Hvorfor barmhjertighed at dræbe vilde dyr er så kontroversieltHvalrossen Freya blev dræbt i Oslo, efter at embedsmænd besluttede, at hun udgjorde en trussel mod mennesker. Kredit:Sheard Photography/Shutterstock To vilde dyr, der forvildede sig fra deres almin
Hvorfor barmhjertighed at dræbe vilde dyr er så kontroversieltHvalrossen Freya blev dræbt i Oslo, efter at embedsmænd besluttede, at hun udgjorde en trussel mod mennesker. Kredit:Sheard Photography/Shutterstock To vilde dyr, der forvildede sig fra deres almin -
 Naturlige enzymer filtrerer hormonforstyrrende kemikalier fra spildevandEt nyt filtreringssystem bruger naturlige molekyler til at fjerne 95 % af hormonpåvirkende kemikalier fra spildevand. Kredit:CC0 Et nyt filtreringssystem, der bruger naturlige molekyler til at fje
Naturlige enzymer filtrerer hormonforstyrrende kemikalier fra spildevandEt nyt filtreringssystem bruger naturlige molekyler til at fjerne 95 % af hormonpåvirkende kemikalier fra spildevand. Kredit:CC0 Et nyt filtreringssystem, der bruger naturlige molekyler til at fje -
 Hvorfor skruer vi ned for radioen, når de gik tabt?Alt, der trækker din opmærksomhed væk fra vejen i mere end to sekunder, betragtes nu som en fare. © ViktorCap/iStockphoto I 1930, radioproducenternes sammenslutning lobbyerede, at passagerer på bagsæ
Hvorfor skruer vi ned for radioen, når de gik tabt?Alt, der trækker din opmærksomhed væk fra vejen i mere end to sekunder, betragtes nu som en fare. © ViktorCap/iStockphoto I 1930, radioproducenternes sammenslutning lobbyerede, at passagerer på bagsæ -
 Syntetisk musefoster med hjerne og bankende hjerte dyrket fra stamcellerNaturlige og syntetiske embryoner side om side med hjerte- og hovedfolder farvet i farve. Kredit:M. Zernicka-Goetz Forskere fra University of Cambridge og Caltech har skabt modelmuseembryoner fra s
Syntetisk musefoster med hjerne og bankende hjerte dyrket fra stamcellerNaturlige og syntetiske embryoner side om side med hjerte- og hovedfolder farvet i farve. Kredit:M. Zernicka-Goetz Forskere fra University of Cambridge og Caltech har skabt modelmuseembryoner fra s
- Udvinding af ædle zink fra affaldsaske
- AI gennemført Beethovens ufærdige 10. symfoni. Sådan lyder det
- Krystaller derhjemme til videnskabsprojekter
- Honning, Jeg krympede vakuumkamrene!
- Amerikanerne valgte borgmestre, der bekymrer sig om klimaforandringer
- Hvordan vil klimaændringer stresse elnettet? Tip:Se på dugpunktstemperaturer