


Teknologi, der simulerer komplekse molekylære interaktioner, kan føre til bedre behandlinger for kræft og COVID-19
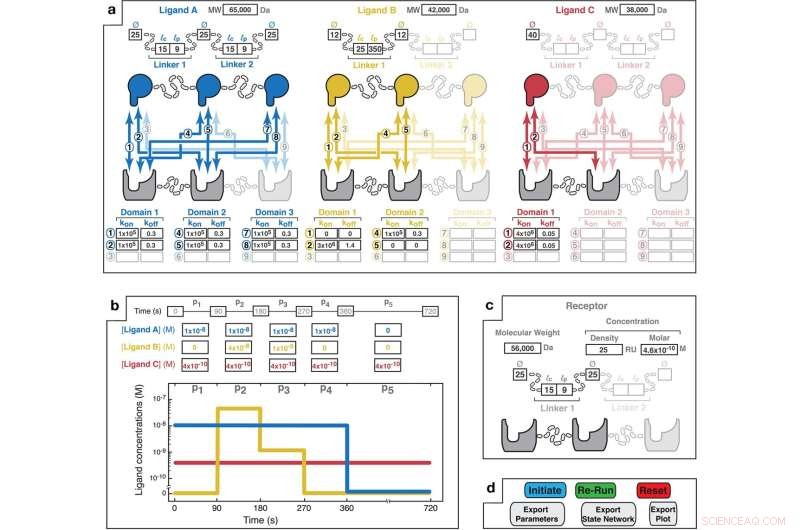
MVsim input design interface giver interaktiv parameterspecifikation for systemer med multivalent, multi-molekylær interaktion. a En peg-og-klik-grænseflade gør det muligt for brugeren at vælge antallet af ligander (op til tre) og valenser af ligand(er) og receptor (op til trivalent), der udgør det multivalente system. Baseret på det valgte design specificerer brugeren strukturen af hver af liganderne ved at indtaste den gældende molekylvægt (MW); bindingsdomænets diametre (Ø); konturlængderne (lc af linkerne (dvs. den maksimale ende-til-ende afstand; f.eks. 3,5 Å og 1,5 Å pr. aminosyre for henholdsvis en tilfældig spole og alfa-helix); og persistenslængderne (lp) af linkere. Yderligere er de anvendelige kombinatoriske interaktioner (nummereret 1 til 9) unikke for hver receptor-ligand-parring fremhævet. Parameterfelter tillader input af monovalente hastighedskonstanter for hver parvis interaktion. Ikke-bindende interaktioner kan angives med k på og k fra værdier på nul (f.eks. som illustreret med ligand B i gult for interaktioner "1" og "5"). b Et inputfelt giver brugeren mulighed for at angive mønstre for de samlede bulkligandkoncentrationer. En associationsfase forekommer i perioder med ikke-nul bulk ligandkoncentration (f.eks. 90-270 s for Ligand B). Dissociationsfaser opstår, når liganden fjernes fra masseopløsningen (f.eks. 360-720 s for Ligand A). Her er ligand C specificeret som kontinuerligt til stede i opløsning i løbet af 720 s af interaktionstidsforløbet. Det grafiske display tillader visualisering af det specificerede bulkkoncentrationspulsmønster. c Brugerinputparametre for receptoren. Receptorkoncentration kan specificeres som enten en SPR-lignende overfladedensitet (målt i RU; hvor 1 RU er lig med ~1 pg/mm 2 ) eller en molær koncentration. Receptortopologi er specificeret i samme form som beskrevet ovenfor for liganderne. d MVsim controller-fanen muliggør initiering, iteration og eksport af bindingssimuleringer. "Initiate" udfører en simulering. "Re-run" udfører en forkortet simulering, der bruges, når der ikke blev foretaget ændringer i systemets valens eller topologi. "Nulstil" genstarter appen og rydder brugerinputparametre fra alle felter. Kredit:Nature Communications (2022). DOI:10.1038/s41467-022-32496-6
Et team ledet af University of Minnesota Twin Cities biomedicinske ingeniører har udviklet en universelt tilgængelig applikation, der kan simulere komplekse molekylære interaktioner, som vil give forskere mulighed for at designe bedre behandlinger for sygdomme som kræft og COVID-19.
Artiklen bygger på en undersøgelse, som forskerne offentliggjorde i 2019. Nu har de udvidet teknologien til at simulere endnu mere komplekse molekylære interaktioner, gjort applikationen nem for ikke-eksperter at bruge og anvendt deres resultater til at kaste lys over, hvordan SARS -CoV-2 virus inficerer kroppen.
Undersøgelsen er publiceret i Nature Communications , og appen, kaldet MVsim, er frit tilgængelig for andre forskere på GitHub.
Simulatoren forudsiger styrken, hastigheden og selektiviteten af multivalente interaktioner, som involverer molekyler, der har flere bindingssteder og kan bruges til at udvikle medicin mod sygdomme, især cancer og COVID-19.
"Multivalente interaktioner er virkelig vigtige i naturlige biologiske systemer, og de begynder nu at blive kreativt udnyttet til at skabe nye terapeutiske lægemidler, der udnytter deres unikke bindingsegenskaber," siger Casim Sarkar, seniorforfatter af papiret og professor ved University of Minnesota Institut for Biomedicinsk Teknik.
"Med multivalente lægemidler kan man i princippet målrette celler meget specifikt på en måde, som ikke er mulig med standard, monovalente lægemidler, men der er mange variabler at tage hensyn til i deres design, og meget af arbejdet på området er gjort indtil nu. gennem eksperimentel forsøg og fejl," tilføjede Sarkar. "Nu ved hjælp af MVsim er vi i stand til at lave gode forudsigelser, der kan bruges til mere rationelt at designe sådanne terapier."
Mange kræftlægemidler binder sig ikke kun til tumorceller, men også til celler, som de ikke er beregnet til at målrette mod, hvilket ofte skaber uønskede bivirkninger for patienten. Ved at optimere specificiteten af multivalente interaktioner ved hjælp af MVsim kan forskere designe lægemidler, der mere specifikt er målrettet mod cellerne i en tumor og samtidig minimerer binding til andre celler i kroppen.
Et andet eksempel er SARS-CoV-2-virus. Forskere ved, at virussen udvikler sig til bedre at inficere vores celler og undgå vores immunsystem, men de molekylære mekanismer bag, hvordan virussen gør dette, er relativt ukendte. Ved at bruge deres MVsim-teknologi var forskere fra University of Minnesota i stand til at udforske denne proces mere i dybden og afdække de hastigheder, hvormed individuelle bindingsdomæner inden for virusets multivalente spidsprotein skifter mellem en celle-inficerende tilstand og en immunundvigende tilstand.
"We essentially have a computational microscope that allows us to look under the hood and see what multivalent proteins such as the SARS-CoV-2 spike protein are doing at a molecular level," Sarkar explained. "This level of molecular detail is hard to capture with a physical experiment. One of the real powers of MVsim is that we can not only learn more about how these systems work but we can also use this tool to design new multivalent interactions for diseases like cancer and COVID-19."
The researchers have already identified potential ways to limit the infectivity of current and future SARS-CoV-2 variants, which they plan to test soon. + Udforsk yderligere
Konstrueret multivalent selvsamlet bindeprotein mod SARS-CoV-2 RBD
 Varme artikler
Varme artikler
-
 UFO psykologiLaramie, Wyoming, psykolog R. Leo Sprinkle sponsorerer en årlig konference på University of Wyoming, hvor kontaktpersoner samles for at dele oplevelser. Sprinkles interesse er mere end akademisk:Han m
UFO psykologiLaramie, Wyoming, psykolog R. Leo Sprinkle sponsorerer en årlig konference på University of Wyoming, hvor kontaktpersoner samles for at dele oplevelser. Sprinkles interesse er mere end akademisk:Han m -
 Intra-row ukrudt mulig med visionsystemerKredit:Wageningen University Forskere ved Wageningen University &Research, BU drivhusbrug, udviklet ukrudtsmaskiner, der er i stand til at udføre lukning inden for rækken. Vores eksperter inden fo
Intra-row ukrudt mulig med visionsystemerKredit:Wageningen University Forskere ved Wageningen University &Research, BU drivhusbrug, udviklet ukrudtsmaskiner, der er i stand til at udføre lukning inden for rækken. Vores eksperter inden fo -
 Neanderthal -DNA ændrede den måde, moderne mennesker ser ud påNår mennesker blandede sig med neandertalere, det påvirkede menneskers risiko for sygdom og endda fysisk udseende. martinwimmer/Getty Images Ordet neandertal bruges ofte som en stenografi for idiot.
Neanderthal -DNA ændrede den måde, moderne mennesker ser ud påNår mennesker blandede sig med neandertalere, det påvirkede menneskers risiko for sygdom og endda fysisk udseende. martinwimmer/Getty Images Ordet neandertal bruges ofte som en stenografi for idiot. -
 Kunne genetisk forbedring i sidste ende gøre menneskeheden dummere?Kan rod med vores eget DNA påvirke menneskeheden negativt? RubberBall Productions/Agency Collection/Getty Images Ingen andre arter på planeten kan matche menneskers evne til at lære, problemløsning o
Kunne genetisk forbedring i sidste ende gøre menneskeheden dummere?Kan rod med vores eget DNA påvirke menneskeheden negativt? RubberBall Productions/Agency Collection/Getty Images Ingen andre arter på planeten kan matche menneskers evne til at lære, problemløsning o
- Hvordan kan du sikkerhedskopiere dine solpaneler?
- Ny proces fremmer området for kulstofudnyttelse
- SpaceX opsender 10 flere Iridium Communications-satellitter
- Mikrober, der lever i en giftig vulkansø, kan indeholde spor til livet på Mars
- Santos-aktierne stiger efter Harbor Energy-overtagelsestilbuddet
- Hubble fastholder en mærkelig exoplanet med fjerntliggende kredsløb, der opfører sig som den læn…