


Genetiske analyser afslører nye vira i horisonten
Pludselig dukker de op, og ligesom SARS-CoV-2 coronavirus kan de udløse store epidemier:Virus, som ingen havde på deres radar. De er ikke rigtig nye, men de har ændret sig genetisk. Især kan udvekslingen af genetisk materiale mellem forskellige virusarter føre til pludselig fremkomst af truende patogener med væsentligt ændrede karakteristika.
Det antyder aktuelle genetiske analyser udført af et internationalt hold af forskere. Virologer fra det tyske cancerforskningscenter (DKFZ) stod for den storstilede undersøgelse, offentliggjort i tidsskriftet PLOS Pathogens .
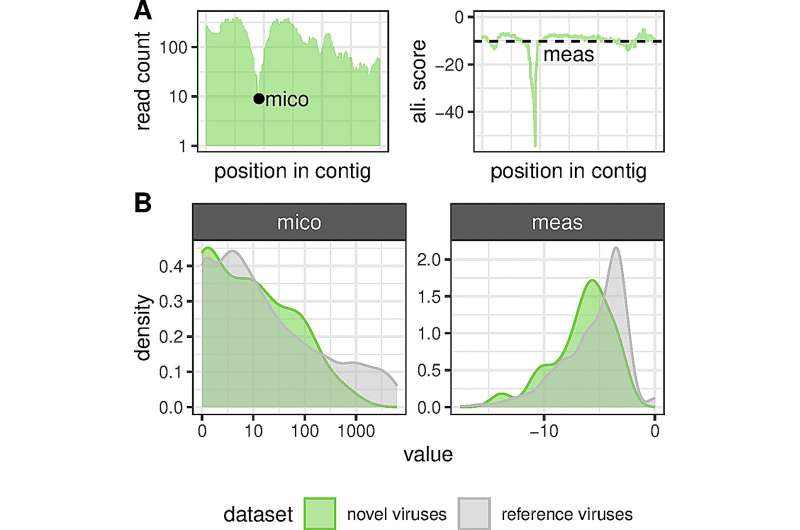
"Ved hjælp af en ny computerstøttet analysemetode opdagede vi 40 hidtil ukendte nidovirus i forskellige hvirveldyr fra fisk til gnavere, herunder 13 coronavirus," rapporterer DKFZ-gruppeleder Stefan Seitz. Ved hjælp af højtydende computere har forskerholdet, som også omfatter Chris Laubers arbejdsgruppe fra Helmholtz Center for Infection Research i Hannover, gennemsøgt næsten 300.000 datasæt. Ifølge virolog Seitz åbner det faktum, at vi nu kan analysere så store mængder data på én gang, helt nye perspektiver.
Virusforskning er stadig i sin relative vorden. Kun en brøkdel af alle vira, der forekommer i naturen, er kendt, især dem, der forårsager sygdomme hos mennesker, husdyr og afgrøder. Den nye metode lover derfor et kvantespring i viden med hensyn til det naturlige virusreservoir. Stefan Seitz og hans kolleger sendte genetiske data fra hvirveldyr gemt i videnskabelige databaser gennem deres højtydende computere med nye spørgsmål. De søgte efter virusinficerede dyr for at opnå og studere viralt genetisk materiale i stor skala. Hovedfokus var på såkaldte nidovirus, som omfatter coronavirus-familien.
Nidovirus, hvis arvemateriale består af RNA (ribonukleinsyre), er udbredt hos hvirveldyr. Denne artsrige gruppe af vira har nogle fælles karakteristika, der adskiller dem fra alle andre RNA-vira og dokumenterer deres forhold. Ellers er nidovirus meget forskellige fra hinanden, dvs. hvad angår størrelsen af deres genom.
En opdagelse er særlig interessant med hensyn til fremkomsten af nye vira:Hos værtsdyr, der samtidigt er inficeret med forskellige vira, kan en rekombination af virale gener forekomme under virusreplikation.
"Tilsyneladende udveksler de nidovirus, vi opdagede i fisk, ofte genetisk materiale mellem forskellige virusarter, selv på tværs af familiegrænser," siger Seitz. Og når fjerne slægtninge "krydser", kan dette føre til fremkomsten af vira med helt nye egenskaber. Ifølge Seitz kan sådanne evolutionære spring påvirke virussenes aggressivitet og farlighed, men også deres tilknytning til visse værtsdyr.
"En genetisk udveksling, som vi har fundet i fiskevirus, vil sandsynligvis også forekomme i pattedyrsvirus," forklarer Seitz. Flagermus, der - ligesom spidsmus - ofte er inficeret med et stort antal forskellige vira, betragtes som en sand smeltedigel. SARS-CoV-2 coronavirus udviklede sig sandsynligvis også hos flagermus og sprang derfra til mennesker.
Efter genudveksling mellem nidovirus ændres det spikeprotein, som viraene sætter sig fast på deres værtsceller med, ofte. Chris Lauber, førsteforfatter af undersøgelsen, kunne vise dette ved hjælp af stamtræsanalyser. Ændring af dette ankermolekyle kan ændre viraenes egenskaber væsentligt til deres fordel – ved at øge deres smitsomhed eller sætte dem i stand til at skifte vært.
Et værtsskifte, især fra dyr til mennesker, kan i høj grad lette spredningen af virussen, hvilket corona-pandemien eftertrykkeligt har vist. Virale "game-changers" kan pludselig dukke op når som helst, blive en massiv trussel, og - hvis push kommer til at skubbe - udløse en pandemi. Udgangspunktet kan være et enkelt dobbelt-inficeret værtsdyr.
Den nye højtydende computerproces kan hjælpe med at forhindre spredning af nye vira. Det muliggør en systematisk søgning efter virusvarianter, der er potentielt farlige for mennesker, forklarer Seitz.
DKFZ-forskeren ser en anden vigtig mulig anvendelse med hensyn til sit særlige forskningsfelt, virusassocieret carcinogenese:"Jeg kunne forestille mig, at vi kunne bruge den nye High Performance Computing (HPC) til systematisk at undersøge kræftpatienter eller immunsvækkede mennesker for virus. Vi ved, at kræft kan udløses af vira, det mest kendte eksempel er humane papillomavira. Men vi ser nok kun toppen af isbjerget indtil videre menneskelig organisme og øge risikoen for ondartede tumorer."
Flere oplysninger: Chris Lauber et al., Deep mining of the Sequence Read Archive afslører store genetiske innovationer i coronavirus og andre nidovirus fra vandlevende hvirveldyr, PLOS Pathogens (2024). DOI:10.1371/journal.ppat.1012163
Journaloplysninger: PLoS-patogener
Leveret af German Cancer Research Center
 Varme artikler
Varme artikler
-
 Kortlægning af den globale indvirkning af krympende gletschere på flodens hvirvelløse dyrFlodens hvirvelløse dyr. Kredit:Liam Marsh, liammarsh.com Flodens hvirvelløse dyr reagerer på samme måde til faldende gletscherdækning, uanset hvor i verden de er, siger forskere, der har vurderet
Kortlægning af den globale indvirkning af krympende gletschere på flodens hvirvelløse dyrFlodens hvirvelløse dyr. Kredit:Liam Marsh, liammarsh.com Flodens hvirvelløse dyr reagerer på samme måde til faldende gletscherdækning, uanset hvor i verden de er, siger forskere, der har vurderet -
 Måling af temperaturen inde i cellerTemperaturændring i brune fedtceller:Mitokondrier og lipiddråber. Kredit:POSTECH Organeller inde i celler tjener konstant specifikke funktioner på samme måde, som individuelle afdelinger varetager
Måling af temperaturen inde i cellerTemperaturændring i brune fedtceller:Mitokondrier og lipiddråber. Kredit:POSTECH Organeller inde i celler tjener konstant specifikke funktioner på samme måde, som individuelle afdelinger varetager -
 Kan en snyltehveps redde dine frugtafgrøder?Forskel i kropsfarve hos voksne vildtype- og ibenholt-mutante fluer. Repræsentative billeder af den dorsale thorax af voksne fluer:OR (A) og e 1 mutant (B). (A) En blå firkant angiver området af inter
Kan en snyltehveps redde dine frugtafgrøder?Forskel i kropsfarve hos voksne vildtype- og ibenholt-mutante fluer. Repræsentative billeder af den dorsale thorax af voksne fluer:OR (A) og e 1 mutant (B). (A) En blå firkant angiver området af inter -
 Nationsskabende eller naturødelæggende? Hvorfor dets tid NZ stod over for miljøskaden fra sin kol…Kredit:National Library of New Zealand, CC BY-NC-ND Måden, hvorpå New Zealand husker europæisk kolonisering, har ændret sig markant i de senere år. Kritikere har skåret væk på det offentlige billed
Nationsskabende eller naturødelæggende? Hvorfor dets tid NZ stod over for miljøskaden fra sin kol…Kredit:National Library of New Zealand, CC BY-NC-ND Måden, hvorpå New Zealand husker europæisk kolonisering, har ændret sig markant i de senere år. Kritikere har skåret væk på det offentlige billed
- Forskere låser spor til et dramatisk kapitel i Jordens geologiske historie
- Ny indsigt i bakterielle toksiner
- Hvordan hjælper kolibrier med pollination?
- Perus rigelige ruiner føler presset af urbanisering
- Er vi alene? Nobelprisen går til tre, der tacklede kosmisk forespørgsel
- Hvordan overbevise folk om at stoppe forurening