


En game changer:Metagenomisk clustering drevet af supercomputere
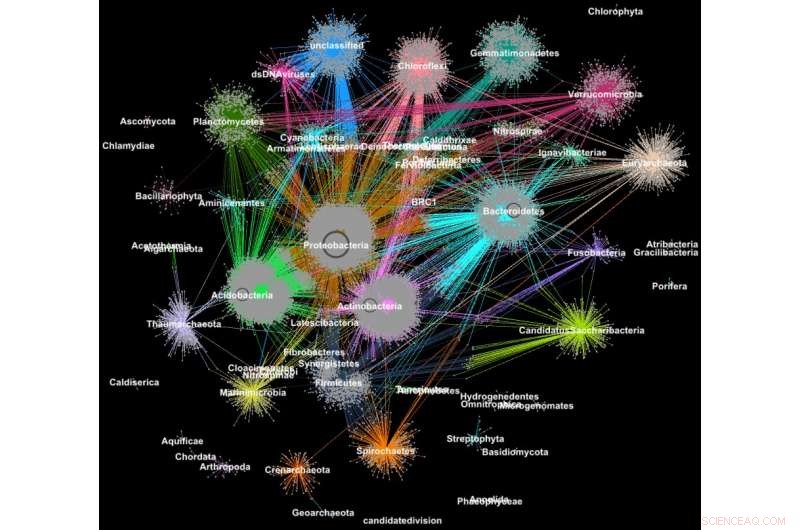
Proteiner fra metagenomer grupperet i familier i henhold til deres taksonomiske klassifikation. Kredit:Georgios Pavlopoulos og Nikos Kyrpides, JGI/Berkeley Lab
Vidste du, at de værktøjer, der bruges til at analysere relationer mellem sociale netværksbrugere eller rangordne websider, også kan være ekstremt værdifulde for at give mening i store videnskabelige data? På et socialt netværk som Facebook, hver bruger (person eller organisation) er repræsenteret som en node, og forbindelserne (relationer og interaktioner) mellem dem kaldes kanter. Ved at analysere disse sammenhænge, forskere kan lære meget om hver enkelt bruger – interesser, hobbyer, indkøbsvaner, venner, etc.
I biologi, lignende graf-clustering algoritmer kan bruges til at forstå de proteiner, der udfører de fleste af livets funktioner. Det anslås, at den menneskelige krop alene indeholder omkring 100, 000 forskellige proteintyper, og næsten alle biologiske opgaver - fra fordøjelse til immunitet - opstår, når disse mikroorganismer interagerer med hinanden. En bedre forståelse af disse netværk kan hjælpe forskere med at bestemme effektiviteten af et lægemiddel eller identificere potentielle behandlinger for en række sygdomme.
I dag, avancerede high-throughput-teknologier gør det muligt for forskere at fange hundredvis af millioner af proteiner, gener og andre cellulære komponenter på én gang og under en række miljøforhold. Klyngealgoritmer anvendes derefter på disse datasæt for at identificere mønstre og relationer, der kan pege på strukturelle og funktionelle ligheder. Selvom disse teknikker har været udbredt i mere end et årti, de kan ikke følge med den strøm af biologiske data, der genereres af næste generations sequencere og mikroarrays. Faktisk, meget få eksisterende algoritmer kan gruppere et biologisk netværk indeholdende millioner af noder (proteiner) og kanter (forbindelser).
Det er grunden til, at et team af forskere fra Department of Energy's (DOE's) Lawrence Berkeley National Laboratory (Berkeley Lab) og Joint Genome Institute (JGI) tog en af de mest populære klyngetilgange i moderne biologi - Markov Clustering (MCL) algoritmen - og ændrede det til at køre hurtigt, effektivt og i stor skala på supercomputere med distribueret hukommelse. I en testsag, deres højtydende algoritme - kaldet HipMCL - opnåede en tidligere umulig bedrift:at samle et stort biologisk netværk indeholdende omkring 70 millioner noder og 68 milliarder kanter på et par timer, bruger cirka 140, 000 processorkerner på National Energy Research Scientific Computing Centers (NERSC) Cori-supercomputer. Et papir, der beskriver dette arbejde, blev for nylig offentliggjort i tidsskriftet Nukleinsyreforskning .
"Den virkelige fordel ved HipMCL er dens evne til at klynge massive biologiske netværk, som var umulige at klynge sammen med den eksisterende MCL-software, således at vi kan identificere og karakterisere det nye funktionelle rum, der er til stede i de mikrobielle samfund, " siger Nikos Kyrpides, som leder JGI's Microbiome Data Science-indsats og Prokaryote Super Program og er medforfatter på papiret. "Desuden kan vi gøre det uden at ofre noget af følsomheden eller nøjagtigheden af den originale metode, hvilket altid er den største udfordring i denne form for skaleringsbestræbelser."
"Efterhånden som vores data vokser, det bliver endnu mere bydende nødvendigt, at vi flytter vores værktøjer ind i højtydende computermiljøer, " tilføjer han. "Hvis du skulle spørge mig, hvor stort er proteinrummet? Sandheden er, vi ved det ikke rigtigt, for indtil nu havde vi ikke de beregningsmæssige værktøjer til effektivt at gruppere alle vores genomiske data og undersøge det funktionelle mørke stof."
Ud over fremskridt inden for dataindsamlingsteknologi, forskere vælger i stigende grad at dele deres data i fællesskabsdatabaser som systemet Integrated Microbial Genomes &Microbiomes (IMG/M), som blev udviklet gennem et årtier gammelt samarbejde mellem forskere ved JGI og Berkeley Labs Computational Research Division (CRD). Men ved at give brugerne mulighed for at lave sammenlignende analyser og udforske mikrobielle samfunds funktionelle muligheder baseret på deres metagenomiske sekvens, fællesskabsværktøjer som IMG/M bidrager også til dataeksplosionen inden for teknologi.
Hvordan tilfældige gåture fører til computerflaskehalse
For at få fat i denne strøm af data, forskere er afhængige af klyngeanalyse, eller klyngedannelse. Dette er i bund og grund opgaven med at gruppere objekter, så elementer i den samme gruppe (klynge) er mere ens end dem i andre klynger. I mere end et årti, beregningsbiologer har favoriseret MCL til at klynge proteiner ved ligheder og interaktioner.
"En af grundene til, at MCL har været populær blandt beregningsbiologer, er, at det er relativt parameterfrit; brugerne behøver ikke at indstille et væld af parametre for at få nøjagtige resultater, og det er bemærkelsesværdigt stabilt over for små ændringer i dataene. Dette er vigtigt, fordi du måske skal omdefinere en lighed mellem datapunkter, eller du skal muligvis korrigere for en lille målefejl i dine data. I disse tilfælde, du ønsker ikke, at dine ændringer ændrer analysen fra 10 klynger til 1, 000 klynger, " siger Aydin Buluç, en CRD-forsker og en af artiklens medforfattere.
Men, tilføjer han, det computerbiologiske samfund støder på en computerflaskehals, fordi værktøjet for det meste kører på en enkelt computerknude, er beregningsmæssigt dyr at udføre og har et stort hukommelsesfodaftryk - som alle begrænser mængden af data, som denne algoritme kan gruppere.
Et af de mest beregnings- og hukommelsesintensive trin i denne analyse er en proces kaldet random walk. Denne teknik kvantificerer styrken af en forbindelse mellem noder, hvilket er nyttigt til at klassificere og forudsige links i et netværk. I tilfælde af en internetsøgning, Dette kan hjælpe dig med at finde et billigt hotelværelse i San Francisco til spring break og endda fortælle dig det bedste tidspunkt at reservere det på. I biologi, et sådant værktøj kan hjælpe dig med at identificere proteiner, der hjælper din krop med at bekæmpe en influenzavirus.
Givet en vilkårlig graf eller netværk, det er svært at kende den mest effektive måde at besøge alle noder og links på. En tilfældig gåtur får en fornemmelse af fodaftrykket ved at udforske hele grafen tilfældigt; den starter ved en knude og bevæger sig vilkårligt langs en kant til en naboknude. Denne proces fortsætter, indtil alle noder på grafnetværket er nået. Fordi der er mange forskellige måder at rejse mellem noder i et netværk, dette trin gentages adskillige gange. Algoritmer som MCL vil fortsætte med at køre denne tilfældige gang-proces, indtil der ikke længere er en signifikant forskel mellem iterationerne.
I ethvert givet netværk, du har muligvis en node, der er forbundet til hundredvis af noder, og en anden node med kun én forbindelse. De tilfældige ture vil fange de stærkt forbundne noder, fordi en anden sti vil blive detekteret, hver gang processen køres. Med disse oplysninger, algoritmen kan med en vis sikkerhed forudsige, hvordan en node på netværket er forbundet med en anden. Mellem hver tilfældige gåtur, Algoritmen markerer sin forudsigelse for hver knude på grafen i en søjle af en Markov-matrix - lidt ligesom en hovedbog - og endelige klynger afsløres i slutningen. Det lyder simpelt nok, men for proteinnetværk med millioner af noder og milliarder af kanter, dette kan blive et ekstremt beregnings- og hukommelsesintensivt problem. Med HipMCL, Berkeley Labs dataloger brugte banebrydende matematiske værktøjer til at overvinde disse begrænsninger.
"Vi har især holdt MCL-rygraden intakt, gør HipMCL til en massivt parallel implementering af den originale MCL-algoritme, " siger Ariful Azad, en datalog i CRD og hovedforfatter af papiret.
Selvom der tidligere har været forsøg på at parallelisere MCL-algoritmen til at køre på en enkelt GPU, værktøjet kunne stadig kun klynge relativt små netværk på grund af hukommelsesbegrænsninger på en GPU, Azad noter.
"Med HipMCL omarbejder vi i det væsentlige MCL-algoritmerne for at køre effektivt, parallelt på tusindvis af processorer, og konfigurer den til at drage fordel af den samlede hukommelse, der er tilgængelig i alle computerknudepunkter, " tilføjer han. "Den hidtil usete skalerbarhed af HipMCL kommer fra dets brug af state-of-the-art algoritmer til sparsom matrix manipulation."
Ifølge Buluç, at udføre en tilfældig gåtur samtidigt fra mange knudepunkter i grafen beregnes bedst ved hjælp af sparse-matrix matrix multiplikation, som er en af de mest basale operationer i den nyligt udgivne GraphBLAS-standard. Buluç og Azad udviklede nogle af de mest skalerbare parallelle algoritmer til GraphBLAS' sparse-matrix matrix multiplikation og modificerede en af deres state-of-the-art algoritmer til HipMCL.
"Kruxet her var at finde den rette balance mellem parallelisme og hukommelsesforbrug. HipMCL udtrækker dynamisk så meget parallelisme som muligt givet den tilgængelige hukommelse, der er allokeret til det, " siger Buluç.
HipMCL:Clustering i skala
Ud over de matematiske innovationer, en anden fordel ved HipMCL er dens evne til at køre problemfrit på ethvert system – inklusive bærbare computere, arbejdsstationer og store supercomputere. Det opnåede forskerne ved at udvikle deres værktøjer i C++ og bruge standard MPI- og OpenMP-biblioteker.
"Vi testede HipMCL grundigt på Intel Haswell, Ivy Bridge og Knights Landing processorer hos NERSC, bruger en op til 2, 000 noder og en halv million tråde på alle processorer, og i alle disse kørsler har HipMCL med succes klynget netværk omfattende tusinder til milliarder af kanter, " siger Buluç. "Vi ser, at der ikke er nogen barriere i antallet af processorer, som den kan bruge til at køre og finder ud af, at den kan klynge netværk 1, 000 gange hurtigere end den originale MCL-algoritme."
"HipMCL kommer til at være virkelig transformerende for beregningsbiologi af big data, ligesom IMG- og IMG/M-systemerne har været for mikrobiom genomik, " siger Kyrpides. "Denne præstation er et vidnesbyrd om fordelene ved tværfagligt samarbejde på Berkeley Lab. Som biologer forstår vi videnskaben, men det har været så uvurderligt at kunne samarbejde med dataloger, der kan hjælpe os med at tackle vores begrænsninger og drive os fremad."
Deres næste skridt er at fortsætte med at omarbejde HipMCL og andre beregningsbiologiske værktøjer til fremtidige exascale-systemer, som vil være i stand til at beregne kvintillionberegninger i sekundet. Dette vil være essentielt, da genomiske data fortsætter med at vokse med en forbløffende hastighed - fordobles omkring hver femte til sjette måned. Dette vil blive gjort som en del af DOE Exascale Computing Projects Exagraph co-design center.
 Varme artikler
Varme artikler
-
 Air France tilbyder at få fat i det franske flyselskab, da tusindvis er strandetAigle Zur, som beskæftiger næsten 1, 200 ansatte, indgav konkursbegæring og indstillede flyvninger i sidste uge Det franske flag luftfartsselskab Air France tilbød mandag at få fat i sin meget min
Air France tilbyder at få fat i det franske flyselskab, da tusindvis er strandetAigle Zur, som beskæftiger næsten 1, 200 ansatte, indgav konkursbegæring og indstillede flyvninger i sidste uge Det franske flag luftfartsselskab Air France tilbød mandag at få fat i sin meget min -
 Træ kan du lide at genbruge beton?Kredit:CC0 Public Domain Forskere ved Institut for Industrividenskab, en del af University of Tokyo, har udviklet en ny procedure for genanvendelse af beton med tilføjelse af kasseret træ. De fand
Træ kan du lide at genbruge beton?Kredit:CC0 Public Domain Forskere ved Institut for Industrividenskab, en del af University of Tokyo, har udviklet en ny procedure for genanvendelse af beton med tilføjelse af kasseret træ. De fand -
 Afstemning på en blockchainKredit:CC0 Public Domain Blockchain-konceptet, som kryptovalutaer arbejder på, kan ekstrapoleres til mange andre områder af livet, såsom stemmesystemer, hvor dens uomtvistelige kæde af beslutninge
Afstemning på en blockchainKredit:CC0 Public Domain Blockchain-konceptet, som kryptovalutaer arbejder på, kan ekstrapoleres til mange andre områder af livet, såsom stemmesystemer, hvor dens uomtvistelige kæde af beslutninge -
 At vende blikket mod terroristerneKredit:CC0 Public Domain Det er kendt, at meget terroraktivitet udnytter kraften og umiddelbarheden af online sociale medier og sociale netværksværktøjer til at koordinere dets angreb, samle stø
At vende blikket mod terroristerneKredit:CC0 Public Domain Det er kendt, at meget terroraktivitet udnytter kraften og umiddelbarheden af online sociale medier og sociale netværksværktøjer til at koordinere dets angreb, samle stø
- Ohio -beboere kæmper for at give Lake Erie juridiske rettigheder
- Kanon,
- Sådan beregnes åbningsstørrelse
- Størrelse betyder noget:Quantum dots kunne gøre solpaneler mere effektive
- Mikrorobotter til at ændre den måde, vi arbejder med cellulært materiale på
- Kemiske ingeniører udvikler ny teori til at bygge forbedrede nanomaterialer