


En vellykket fononberegning inden for kvante Monte Carlo-rammen
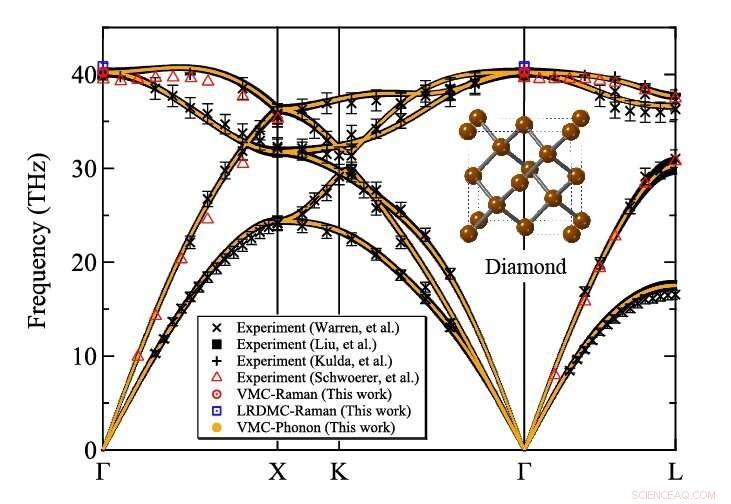
Phonon-spredning af diamant beregnet på det variationelle Monte Carlo-niveau af TurboRVB. Kredit:Kousuke Nakano fra JAIST
Fokus og det ultimative mål for beregningsmæssig forskning inden for materialevidenskab og kondenseret stoffysik er at løse Schrödinger-ligningen - den grundlæggende ligning, der beskriver, hvordan elektroner opfører sig inde i stof - nøjagtigt (uden at ty til forenklede tilnærmelser). Mens eksperimenter bestemt kan give interessant indsigt i et materiales egenskaber, det er ofte beregninger, der afslører den underliggende fysiske mekanisme. Imidlertid, beregninger behøver ikke være afhængige af eksperimentelle data og kan, faktisk, udføres selvstændigt, en tilgang kendt som "ab initio-beregninger." Density functional theory (DFT) er et populært eksempel på en sådan tilgang.
For de fleste materialeforskere og fysikere af kondenseret stof, DFT-beregninger er deres fags brød og smør. Imidlertid, på trods af at det er en kraftfuld teknik, DFT har haft begrænset succes med "stærkt korrelerede materialer" - materialer med usædvanlige elektroniske og magnetiske egenskaber. Disse materialer, mens de er interessante i sig selv, har også teknologiske nyttige egenskaber, et faktum, der stærkt motiverer en ab initio-ramme, der er egnet til at beskrive dem.
Til det formål, en ramme kendt som "ab initio quantum Monte Carlo" (QMC) har vist betydeligt lovende og forventes at blive den næste generation af elektroniske strukturberegninger på grund af dens overlegenhed i forhold til DFT. Imidlertid, selv QMC er stort set begrænset til beregninger af energi og atomkræfter, begrænser dets anvendelighed til at beregne nyttige materialeegenskaber.
Nu, i en banebrydende undersøgelse offentliggjort i Fysisk gennemgang B (Redaktørens forslag), videnskabsmænd har taget tingene til det næste niveau baseret på en tilgang, der giver dem mulighed for at reducere den statistiske fejl i atomkraftevaluering med to størrelsesordener og efterfølgende fremskynde beregningen med en faktor på 10 4 ! "Den drastiske reduktion i beregningstid vil i høj grad udvide rækken af QMC-beregninger og muliggøre meget nøjagtig forudsigelse af atomare egenskaber af materialer, der har været svære at håndtere, " bemærker adjunkt Kousuke Nakano fra Japan Advanced Institute of Science and Technology (JAIST), WHO, sammen med sine kolleger Prof. Ryo Maezono fra JAIST, Prof. Sandro Sorella fra International School for Advanced Studies (SISSA), Italien, og Dr. Tommaso Morresi og Prof. Michele Casula fra Sorbonne Université, Frankrig, ledet denne banebrydende præstation.
Holdet anvendte deres udviklede metode til at beregne atomvibrationer af diamant, et typisk referencemateriale, som et proof-of-concept og viste, at resultaterne var i overensstemmelse med eksperimentelle værdier. For at udføre disse beregninger, de brugte en stor computer, Cray-XC40, placeret på Research Center for Advanced Computing Infrastructure ved Japan Advanced Institute of Science and Technology (JAIST), Japan, sammen med en anden beliggende på RIKEN, Japan. Holdet gjorde brug af en QMC-softwarepakke kaldet "TurboRVB, " oprindeligt lanceret af prof. Sorella og prof. Casula og senere udviklet af prof. Nakano sammen med andre, at udføre fononspredningsberegninger for diamanter, der tidligere var utilgængelige, udvider dets omfang kraftigt.
Prof. Nakano ser frem til anvendelsen af QMC i materialeinformatik (MI), et felt dedikeret til design og søgning efter nye materialer ved hjælp af teknikker inden for informationsvidenskab og beregningsfysik. "Mens MI i øjeblikket er styret af DFT, den hurtige udvikling i computerens ydeevne, såsom exascale supercomputeren, vil hjælpe QMC med at vinde popularitet. I den forbindelse vores udviklede metode vil være meget nyttig til at designe nye materialer med virkelige applikationer, " konkluderer en optimistisk Dr. Nakano.
 Varme artikler
Varme artikler
-
 Stealth -ark skjuler varme genstande fra nysgerrige infrarøde øjneEt nyudviklet stealth -ark kan skjule varme genstande som menneskelige kroppe eller militære køretøjer fra infrarøde kameraer. Kredit:Hongrui Jiang Infrarøde kameraer er de varmefølende øjne, der
Stealth -ark skjuler varme genstande fra nysgerrige infrarøde øjneEt nyudviklet stealth -ark kan skjule varme genstande som menneskelige kroppe eller militære køretøjer fra infrarøde kameraer. Kredit:Hongrui Jiang Infrarøde kameraer er de varmefølende øjne, der -
 5 ting, du ikke vidste om obduktionerRetsmedicineren Dr. Bennet Omalu diskuterer et diagram over hans obduktion af Stephon Clark, der blev dræbt af to Sacramento -politifolk i 2018. Clarks familie anmodede om en uafhængig obduktion. Just
5 ting, du ikke vidste om obduktionerRetsmedicineren Dr. Bennet Omalu diskuterer et diagram over hans obduktion af Stephon Clark, der blev dræbt af to Sacramento -politifolk i 2018. Clarks familie anmodede om en uafhængig obduktion. Just -
 Forskere forsøger at afdække, hvad der gør Stradivarius violiner specielle – men spilder de deres…Kredit:Kerinin/Flickr, CC BY-SA Stradivarius violiner er kendt for deres angiveligt overlegne lyd sammenlignet med andre instrumenter. Dette har resulteret i adskillige undersøgelser, der leder ef
Forskere forsøger at afdække, hvad der gør Stradivarius violiner specielle – men spilder de deres…Kredit:Kerinin/Flickr, CC BY-SA Stradivarius violiner er kendt for deres angiveligt overlegne lyd sammenlignet med andre instrumenter. Dette har resulteret i adskillige undersøgelser, der leder ef -
 Kemiske og fysiske egenskaber ved stålStål er en legering, en kombination af metal lavet af jern og kulstof. Kulstofindholdet i stål når maksimalt 1,5 procent. På grund af dens hårdhed og styrke bruges stål til opførelse af bygninger, bro
Kemiske og fysiske egenskaber ved stålStål er en legering, en kombination af metal lavet af jern og kulstof. Kulstofindholdet i stål når maksimalt 1,5 procent. På grund af dens hårdhed og styrke bruges stål til opførelse af bygninger, bro
- Hvad er en encellær eukaryote?
- Ny briefing fremhæver den skadelige virkning af COVID-19 på hovedgader
- Sæt farten ned:Reducerede hastighedsgrænser redder liv i travle byer
- Forskere skal spore krystallers reaktion på det elektriske felt
- Den mest nøjagtige måling af sjældent mesonforfald bekræfter moderne fysik
- Sådan konverteres mil til timer