


Molekylær dynamik, maskinlæring skaber hyperprædiktive computermodeller
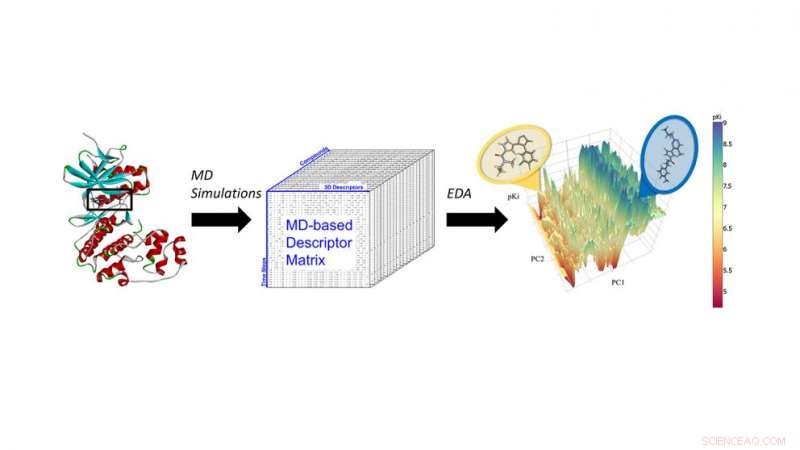
Molecular dynamics (MD) simuleringer af ERK2-hæmmere for at udtrække MD-deskriptorer til næste generations keminformatikanalyse og maskinlæring. Kredit:North Carolina State University
Forskere fra North Carolina State University har vist, at simuleringer af molekylær dynamik og maskinlæringsteknikker kunne integreres for at skabe mere nøjagtige computerforudsigelsesmodeller. Disse "hyperprædiktive" modeller kunne bruges til hurtigt at forudsige, hvilke nye kemiske forbindelser der kunne være lovende lægemiddelkandidater.
Lægemiddeludvikling er en dyr og tidskrævende proces. For at indsnævre antallet af kemiske forbindelser, der kunne være potentielle lægemiddelkandidater, forskere bruger computermodeller, der kan forudsige, hvordan en bestemt kemisk forbindelse kan interagere med et biologisk mål af interesse - f.eks. et nøgleprotein, der kan være involveret i en sygdomsproces. Traditionelt, dette gøres via quantitative structure-activity relationship (QSAR) modellering og molekylær docking, som er afhængige af 2- og 3-D-oplysninger om disse kemikalier.
Denis Fourches, adjunkt i beregningskemi, ønskede at forbedre nøjagtigheden af disse QSAR-modeller. "Når du screener et sæt på 30 millioner forbindelser, du har ikke nødvendigvis brug for en meget høj pålidelighed med din model - du får bare en idé om top 5 eller 10 procent af det virtuelle bibliotek. Men hvis du forsøger at indsnævre et felt på 200 analoger til 10, hvilket er mere almindeligt tilfældet ved udvikling af lægemidler, din modelleringsteknik skal være ekstremt nøjagtig. Nuværende teknikker er bestemt ikke pålidelige nok."
Fourches og Jeremy Ash, en kandidatstuderende i bioinformatik, besluttede at inkorporere resultaterne af molekylær dynamikberegninger - alle-atom-simuleringer af, hvordan en bestemt forbindelse bevæger sig i bindingslommen på et protein - i forudsigelsesmodeller baseret på maskinlæring.
"De fleste modeller bruger kun de todimensionelle strukturer af molekyler, " siger Fourches. "Men i virkeligheden, kemikalier er komplekse tredimensionelle genstande, der bevæger sig, vibrerer og har dynamiske intermolekylære interaktioner med proteinet, når det først er forankret i dets bindingssted. Det kan du ikke se, hvis du bare ser på 2-D eller 3-D strukturen af et givet molekyle."
I en proof-of-concept undersøgelse, Fourches og Ash så på ERK2-kinasen - et enzym forbundet med flere typer kræft - og en gruppe på 87 kendte ERK2-hæmmere, lige fra meget aktiv til inaktiv. De kørte uafhængige simuleringer af molekylær dynamik (MD) for hver af disse 87 forbindelser og beregnede kritisk information om fleksibiliteten af hver forbindelse én gang i ERK2-lommen. Derefter analyserede de MD-deskriptorerne ved hjælp af keminformatikteknikker og maskinlæring. MD-deskriptorerne var i stand til nøjagtigt at skelne aktive ERK2-hæmmere fra svagt aktive og inaktive, hvilket ikke var tilfældet, når modellerne kun brugte 2-D og 3-D strukturel information.
"Vi havde allerede data om disse 87 molekyler og deres aktivitet på ERK2, " siger Fourches. "Så vi testede for at se, om vores model var i stand til pålideligt at finde de mest aktive forbindelser. Ja, den skelnede nøjagtigt mellem stærke og svage ERK2-hæmmere, og fordi MD-deskriptorer kodede de interaktioner, som disse forbindelser skaber i lommen på ERK2, det gav os også mere indsigt i, hvorfor de stærke inhibitorer fungerede godt.
"Før fremskridt inden for databehandling tillod os at simulere denne type data, det ville have taget os seks måneder at simulere et enkelt molekyle i lommen på ERK2. Takket være GPU-acceleration, nu tager det kun tre timer. Det er en game changer. Jeg håber, at inkorporering af data udvundet fra molekylær dynamik i QSAR-modeller vil muliggøre en ny generation af hyperprædiktive modeller, der vil hjælpe med at bringe nye, effektive lægemidler på markedet endnu hurtigere. Det er kunstig intelligens, der arbejder for, at vi opdager morgendagens stoffer."
 Varme artikler
Varme artikler
-
 Simpel one-pot syntese af tricykliske peptider, der kan droppesStruktur af vancomycin og et tricyklisk peptidformat inspireret af dets multicykliske struktur. Kredit:HIMS Kemikere ved University of Amsterdams Van t Hoff Institute for Molecular Sciences (HIMS)
Simpel one-pot syntese af tricykliske peptider, der kan droppesStruktur af vancomycin og et tricyklisk peptidformat inspireret af dets multicykliske struktur. Kredit:HIMS Kemikere ved University of Amsterdams Van t Hoff Institute for Molecular Sciences (HIMS) -
 Hvorfor salt i vand kan udføre elektricitetFor at forstå, hvorfor saltvand fører elektricitet, skal vi først forstå, hvad elektricitet er. Elektricitet er en konstant strøm af elektroner eller elektrisk ladede partikler gennem et stof. I nogle
Hvorfor salt i vand kan udføre elektricitetFor at forstå, hvorfor saltvand fører elektricitet, skal vi først forstå, hvad elektricitet er. Elektricitet er en konstant strøm af elektroner eller elektrisk ladede partikler gennem et stof. I nogle -
 At studere virale udbrud i enkeltceller kan afsløre nye måder at besejre dem påKredit:CC0 Public Domain Mange vira, herunder hiv og influenza A, muterer så hurtigt, at identifikation af effektive vacciner eller behandlinger er som at forsøge at ramme et bevægeligt mål. En be
At studere virale udbrud i enkeltceller kan afsløre nye måder at besejre dem påKredit:CC0 Public Domain Mange vira, herunder hiv og influenza A, muterer så hurtigt, at identifikation af effektive vacciner eller behandlinger er som at forsøge at ramme et bevægeligt mål. En be -
 Team fremstiller magneter udelukkende af sjældne jordarter fra USACMI-forsker Ikenna Nlebedim har prøver af neodym-jern-bor-magneter, der er hentet og fremstillet udelukkende i USA. Evnen til at producere sjældne jordarters magneter på hjemmemarkedet kunne give posi
Team fremstiller magneter udelukkende af sjældne jordarter fra USACMI-forsker Ikenna Nlebedim har prøver af neodym-jern-bor-magneter, der er hentet og fremstillet udelukkende i USA. Evnen til at producere sjældne jordarters magneter på hjemmemarkedet kunne give posi
- Den garvede amerikanske pilot Wally Funk for at opfylde rumdrømmen 60 år efter
- Sådan finder du manglende koordinater med Slope
- Er MXenes fremtiden for nanoteknologi?
- Biomedicinsk hudlignende bandage er elastisk, holdbar og langtidsholdbar
- Pennsylvania lader Uber selvkørende biler tilbage på vejene
- Hurtigere, mere præcis lab-on-a-chip holder løfte om tidlig kræftdiagnose