


Computermodel til design af proteinsekvenser, der er optimeret til at binde til lægemiddelmål
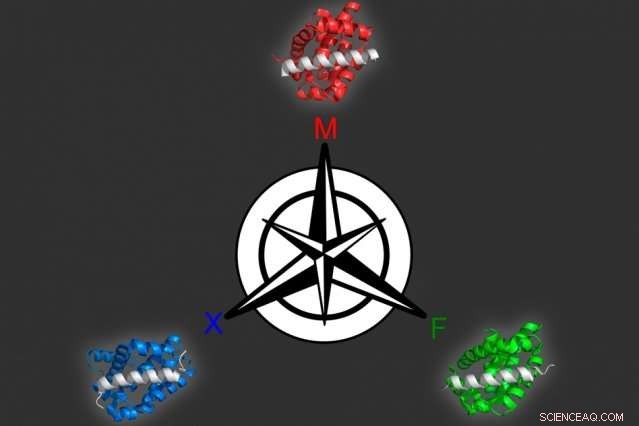
Ved at bruge en computermodelleringstilgang, som de udviklede, MIT-biologer identificerede tre forskellige proteiner, der kan binde selektivt til hver af tre lignende mål, alle medlemmer af Bcl-2-familien af proteiner. Kredit:Vincent Xue
At designe syntetiske proteiner, der kan fungere som lægemidler mod kræft eller andre sygdomme, kan være en kedelig proces:Det involverer generelt at skabe et bibliotek med millioner af proteiner, derefter screening af biblioteket for at finde proteiner, der binder det korrekte mål.
MIT-biologer er nu kommet med en mere raffineret tilgang, hvor de bruger computermodellering til at forudsige, hvordan forskellige proteinsekvenser vil interagere med målet. Denne strategi genererer et større antal kandidater og giver også større kontrol over en række proteinegenskaber, siger Amy Keating, en professor i biologi og biologisk teknik og leder af forskergruppen.
"Vores metode giver dig et meget større spillefelt, hvor du kan vælge løsninger, der er meget forskellige fra hinanden og vil have forskellige styrker og forpligtelser, " siger hun. "Vores håb er, at vi kan levere en bredere vifte af mulige løsninger for at øge gennemstrømningen af disse indledende hits til nyttige, funktionelle molekyler."
I et papir, der vises i Proceedings of the National Academy of Sciences ugen 15. okt. Keating og hendes kolleger brugte denne tilgang til at generere flere peptider, der kan målrette mod forskellige medlemmer af en proteinfamilie kaldet Bcl-2, som er med til at drive kræftvækst.
Nylige ph.d.-modtagere Justin Jenson og Vincent Xue er hovedforfatterne af papiret. Andre forfattere er postdoc Tirtha Mandal, tidligere laboratorietekniker Lindsey Stretz, og tidligere postdoc Lothar Reich.
Modellering af interaktioner
Protein medicin, også kaldet biopharmaceuticals, er en hurtigt voksende klasse af lægemidler, der lover at behandle en lang række sygdomme. Den sædvanlige metode til at identificere sådanne lægemidler er at screene millioner af proteiner, enten tilfældigt valgt eller udvalgt ved at skabe varianter af proteinsekvenser, der allerede har vist sig at være lovende kandidater. Dette involverer konstruktion af vira eller gær til at producere hver af proteinerne, derefter udsætte dem for målet for at se, hvilke der binder bedst.
"Det er standardtilgangen:Enten helt tilfældigt, eller med en vis forhåndsviden, designe et bibliotek af proteiner, og så ud og fiske i biblioteket for at trække de mest lovende medlemmer ud, " siger Keating.
Selvom metoden fungerer godt, det producerer normalt proteiner, der kun er optimeret til et enkelt træk:hvor godt det binder til målet. Det giver ikke mulighed for kontrol over andre funktioner, der kunne være nyttige, såsom egenskaber, der bidrager til et proteins evne til at trænge ind i celler eller dets tendens til at fremkalde et immunrespons.
"Der er ingen indlysende måde at gøre den slags ting på - specificer et positivt ladet peptid, for eksempel - ved at bruge brute force-biblioteksscreeningen, " siger Keating.
Et andet ønskeligt træk er evnen til at identificere proteiner, der binder tæt til deres mål, men ikke til lignende mål, som er med til at sikre, at lægemidler ikke får utilsigtede bivirkninger. Standardtilgangen tillader forskere at gøre dette, men eksperimenterne bliver mere besværlige, siger Keating.
Den nye strategi involverer først at skabe en computermodel, der kan relatere peptidsekvenser til deres bindingsaffinitet for målproteinet. For at skabe denne model, forskerne valgte først omkring 10, 000 peptider, hver 23 aminosyrer i længden og spiralformet struktur, og testede deres binding til tre forskellige medlemmer af Bcl-2-familien. De valgte med vilje nogle sekvenser, de allerede vidste ville binde godt, plus andre de vidste ikke ville, så modellen kunne inkorporere data om en række bindingsevner.
Fra dette sæt af data, modellen kan producere et "landskab" af, hvordan hver peptidsekvens interagerer med hvert mål. Forskerne kan derefter bruge modellen til at forudsige, hvordan andre sekvenser vil interagere med målene, og generere peptider, der opfylder de ønskede kriterier.
Ved at bruge denne model, forskerne producerede 36 peptider, som blev forudsagt at binde et familiemedlem tæt, men ikke de to andre. Alle kandidaterne klarede sig særdeles godt, da forskerne testede dem eksperimentelt, så de prøvede et mere vanskeligt problem:at identificere proteiner, der binder til to af medlemmerne, men ikke det tredje. Mange af disse proteiner var også vellykkede.
"Denne tilgang repræsenterer et skift fra at stille et meget specifikt problem og derefter designe et eksperiment for at løse det, at investere noget arbejde på forhånd for at skabe dette landskab af, hvordan sekvens er relateret til funktion, at fange landskabet i en model, og derefter være i stand til at udforske det efter behag for flere ejendomme, " siger Keating.
Sagar Khare, en lektor i kemi og kemisk biologi ved Rutgers University, siger, at den nye tilgang er imponerende i sin evne til at skelne mellem tæt beslægtede proteinmål.
"Selektivitet af lægemidler er afgørende for at minimere virkninger uden for målet, og ofte er selektivitet meget vanskelig at kode, fordi der er så mange lignende molekylære konkurrenter, som også vil binde lægemidlet bortset fra det tilsigtede mål. Dette arbejde viser, hvordan man koder denne selektivitet i selve designet, " siger Khare, som ikke var involveret i undersøgelsen. "Anvendelser i udviklingen af terapeutiske peptider vil næsten helt sikkert følge."
Selektive stoffer
Medlemmer af Bcl-2-proteinfamilien spiller en vigtig rolle i reguleringen af programmeret celledød. Dysregulering af disse proteiner kan hæmme celledød, hjælper tumorer med at vokse ukontrolleret, så mange medicinalfirmaer har arbejdet på at udvikle lægemidler, der er målrettet mod denne proteinfamilie. For at sådanne lægemidler skal være effektive, det kan være vigtigt for dem at målrette mod kun ét af proteinerne, fordi at forstyrre dem alle kan forårsage skadelige bivirkninger i raske celler.
"I mange tilfælde, kræftceller ser ud til at bruge kun et eller to medlemmer af familien til at fremme celleoverlevelse, " siger Keating. "Generelt, det er anerkendt, at det ville være meget bedre at have et panel af selektive midler end et råt værktøj, der bare slog dem alle ud."
Forskerne har ansøgt om patenter på de peptider, de identificerede i denne undersøgelse, og de håber, at de vil blive yderligere testet som mulige stoffer. Keatings laboratorium arbejder nu på at anvende denne nye modelleringstilgang til andre proteinmål. Denne form for modellering kunne være nyttig til ikke kun at udvikle potentielle lægemidler, men også generere proteiner til brug i landbrugs- eller energianvendelser, hun siger.
Denne historie er genudgivet med tilladelse fra MIT News (web.mit.edu/newsoffice/), et populært websted, der dækker nyheder om MIT-forskning, innovation og undervisning.
Sidste artikelEn stabiliserende påvirkning muliggør udvikling af lithium-svovlbatterier
Næste artikelCellulært stressforsvar
 Varme artikler
Varme artikler
-
 Undersøgelse udvider, hvad vi ved om naturlige, billige måder at fjerne forurenende stoffer fra va…Inden for timer, tørrede og malede avocadoskaller hjælper med at fjerne methylenblåt farvestof fra vand gennem en proces, der kaldes adsorption. Kredit:Dickinson College Nypubliceret forskning fra
Undersøgelse udvider, hvad vi ved om naturlige, billige måder at fjerne forurenende stoffer fra va…Inden for timer, tørrede og malede avocadoskaller hjælper med at fjerne methylenblåt farvestof fra vand gennem en proces, der kaldes adsorption. Kredit:Dickinson College Nypubliceret forskning fra -
 Thermodynamiske egenskaber ved heveinGummi latex Hevea brasiliensis . Kredit:Lobachevsky University Hevein er et lille protein (4,7 kDa) bestående af 43 aminosyrerester. Det er hovedkomponenten i bundfraktionen af gummitræ ( Hev
Thermodynamiske egenskaber ved heveinGummi latex Hevea brasiliensis . Kredit:Lobachevsky University Hevein er et lille protein (4,7 kDa) bestående af 43 aminosyrerester. Det er hovedkomponenten i bundfraktionen af gummitræ ( Hev -
 Proteinblanding under blåt lysSkema af CRY2PHR klyngemekanismen udløst af blåt lys. I dette optogenetiske system, CRY2-klynger (blå og pink) styres reversibelt af blåt lys. Hvis CRY2 er forbundet med fluorescerende proteiner (grøn
Proteinblanding under blåt lysSkema af CRY2PHR klyngemekanismen udløst af blåt lys. I dette optogenetiske system, CRY2-klynger (blå og pink) styres reversibelt af blåt lys. Hvis CRY2 er forbundet med fluorescerende proteiner (grøn -
 Ny opdagelse kan muliggøre præcis forudsigelse af kræftspredning, før kræft udvikler sigVENSTRE:Spheroid af brystkræftceller, der bryder gennem basalmembranen for at invadere i det omkringliggende område. Denne evne bestemmes af forholdet mellem netrin-molekyler til stede i basalmembrans
Ny opdagelse kan muliggøre præcis forudsigelse af kræftspredning, før kræft udvikler sigVENSTRE:Spheroid af brystkræftceller, der bryder gennem basalmembranen for at invadere i det omkringliggende område. Denne evne bestemmes af forholdet mellem netrin-molekyler til stede i basalmembrans
- Kvinder knuser glasloftet for kun at falde ned af glasklinten
- Canada, hvis Trudeau vinder, at ramme nul -emissioner inden 2050:minister
- Nye beviser viser hurtig reaktion i det vestgrønlandske landskab på arktiske klimaændringer
- Snapchat lancerer egen multi-player gaming platform
- Varmebehandling giver præcis kontrol over katalytisk aktivitet af metalsulfidnanopartikler
- Hvordan big data omformer aldrende forskning og uddannelse