


Supercomputing, neutroner forenes for at optrevle strukturer af iboende forstyrret protein
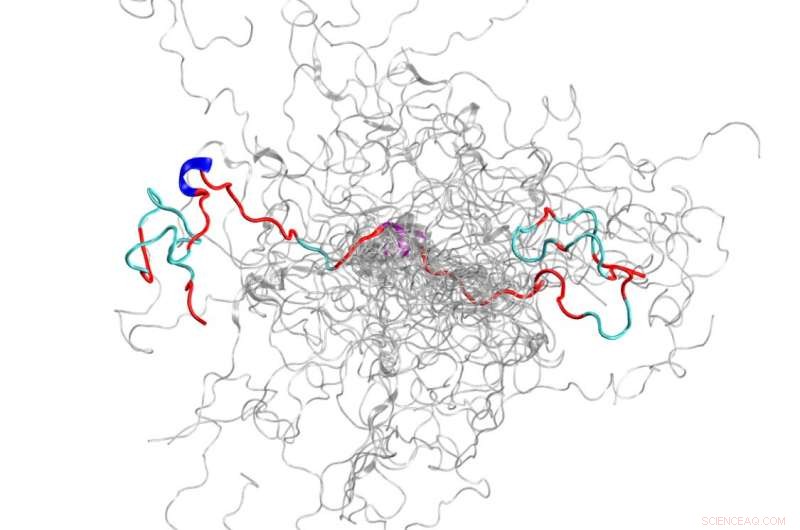
Konfigurationsensemblet (en samling af 3D-strukturer) af et iboende forstyrret protein, N-terminalen af c-Src kinase, som er et vigtigt signalprotein hos mennesker. Kredit:Oak Ridge National Laboratory, US Department of Energy
Ved at bruge Titan-supercomputeren og Spallation Neutron Source ved Department of Energy's Oak Ridge National Laboratory, Forskere har skabt den mest nøjagtige 3D-model til dato af et iboende uordnet protein, afslører ensemblet af dets strukturer på atomniveau.
Som navnet indikerer, en IDP vedtager ikke en beordret, statisk struktur som andre proteiner; i stedet, den er fleksibel og kan anvende flere 3D-strukturer. Denne mangel på en unik struktur er nødvendig for IDP's biologiske funktion, men gør det teknisk udfordrende at studere. IDP'er kan være et helt protein eller et domæne af et ellers struktureret protein, og de udgør en stor del af mennesker, mikrobe, og planteproteiner.
Loukas Petridis, en stabsforsker ved Center for Molekylær Biofysik ved ORNL, har rettet et team af forskere til en ny måde at skabe nøjagtige fysiske modeller af sådanne fleksible biosystemer, hvilket kan føre til en bedre forståelse af deres biologiske funktioner. I løbet af de seneste tre år har holdet har kombineret neutronspredningseksperimenter med forbedrede sampling molekylær dynamik (MD) simuleringer, der er så beregningskrævende, at de krævede behandlingskraften fra Titan, den nyligt nedlagte 27 petaflop Cray XK7 ved Oak Ridge Leadership Computing Facility, en DOE Office of Science User Facility på ORNL.
"At studere disse internt fordrevne er ret svært, fra både eksperimenter og modellering, " sagde Utsab Shrestha, hovedforfatteren af holdets papir, for nylig offentliggjort i Proceedings of the National Academy of Sciences . "Vi tænkte ikke kun på det fra eksperiment eller simulering alene, vi planlagde på en måde, så vi ville synergi begge disse tilgange – kombinere dem på en måde, så vi kunne få mere præcis information om internt fordrevne. Specifikt, simuleringer hjalp os med at generere et nøjagtigt ensemble af IDP ved atomopløsning, hvilket er svært at bestemme ud fra eksperimenter alene."
Typisk, forskere udfører eksperimenter såsom småvinklet neutronspredning, småvinklet røntgenspredning, eller kernemagnetisk resonans til at sondere fleksible biologiske systemer. Imidlertid, disse metoder giver ikke et detaljeret billede på atomniveau af en IDP's 3D-strukturer, kendt som dets konfigurationsensemble. Desuden, de kan kun producere ensemble-gennemsnitsdata, snarere end de specifikke underliggende proteinstrukturkonfigurationer. Forskere har også udført computersimuleringer af IDP og sammenlignet dem med sådanne eksperimenter, i håb om at få de samme resultater for at verificere nøjagtigheden af deres modeller.
"Men de ender med at være uenige i eksperimenterne, " sagde Petridis. "Og på grund af uoverensstemmelsen mellem simuleringerne og eksperimenterne, de skal omvægte simuleringerne – de skal justere simuleringsresultaterne for at få dem til at matche eksperimenterne, hvilket er frustrerende. Det var state of the art indtil vores arbejde."
Computer MD-simuleringer udført af Shrestha brugte forbedrede prøvetagningsmetoder, der lykkedes med at matche ikke kun neutronspredningseksperimenter - udført af Viswanathan Gurumoorthy og hans kolleger hos SNS, en DOE Office of Science User Facility på ORNL - men også tidligere offentliggjorte NMR-data. Disse MD-simuleringer bruger fysik til at bestemme, hvordan proteiner bevæger sig. Nøglen til holdets succes var at køre mange MD-simuleringer parallelt på Titan, giver simuleringerne mulighed for at kommunikere med hinanden og udveksle information.
"Dette er meget vigtigt, fordi det giver simuleringen mulighed for at prøve et større konfigurationsrum, udforske flere af de tredimensionelle strukturer på en mere effektiv måde, " sagde Petridis. "Det er derfor, denne forbedrede prøveudtagnings-MD kan producere resultater, som den normale MD-simulering ikke kan. Vi skulle køre en normal MD-simulering i årevis for at opnå de samme resultater."
IDP'en, som holdet valgte at studere, er det N-terminale domæne af c-Src kinase, som er et vigtigt signalprotein hos mennesker. Mutationer i dette komplekse protein er blevet korreleret med kræft, hvilket også gør det til et vigtigt lægemiddelmål. Mens du kortlægger dette tidligere uklare domæne, forskerne var i stand til at opdage ny information om dens 3D-strukturer, som tidligere metoder ikke havde vist. For eksempel, selvom det stort set er uordnet, dette protein danner forbigående ordnede strukturer, såsom helixer.
"Kombinationen af neutronspredningsforsøg og simulering er meget kraftfuld, " sagde Petridis. "Validering af simuleringerne ved sammenligning med neutronspredningseksperimenter er afgørende for at have tillid til simuleringsresultaterne. De validerede simuleringer kan derefter give detaljerede oplysninger, som ikke er direkte opnået ved eksperimenter."
Den detaljerede computermodel af IDP's 3-D strukturensemble åbner døren til mere eksperimentering. For eksempel, forskere kunne simulere effekten af phosphorylering (tilsætning af en phosphatgruppe til proteinet, der kan regulere proteinets funktion) for at se, hvilke strukturelle ændringer der finder sted i c-Src kinase, som kan påvirke dets funktion. Mutationers rolle kunne også undersøges:Hvis en forsker ændrer en aminosyre i kæden, hvordan påvirker dette strukturen eller ensemblet af strukturer?
"Der er en masse ubesvarede spørgsmål til især c-Src kinase, som kunne besvares med hensyn til interaktioner med andre partnere - effekten af fosforylering, virkningen af mutationer, " sagde Petridis.
Ud over de potentielle videnskabelige anvendelser af selve modellen, Petridis ser muligheder for at anvende brugen af højtydende computing til at køre forbedret sampling MD til at studere strukturerne af mange andre vigtige IDP'er, som kunne give indsigt i deres funktion. Og mere bredt, holdet ønsker at udvikle simuleringsteknologier, der kan reproducere småvinklede neutronspredningsprofiler af endnu mere komplekse biologiske systemer.
"Vi ønsker ikke kun at undersøge de forstyrrede proteiner - vi ønsker at have meget større systemer, der indeholder ordnede og uordnede domæner, der kan interagere med membraner eller DNA, " sagde Petridis. "Neutronspredning er, efter min mening, den bedste eksperimentelle teknik til at sondere disse multi-komponent systemer – f.eks. et protein, der interagerer med en membran eller et protein, der interagerer med DNA. Men, stadig, neutronspredning har brug for de nøjagtige simuleringer for bedre at fortolke dataene."
 Varme artikler
Varme artikler
-
 Forskere identificerer elektroniske og strukturelle dynamikker i katalytiske centre i enkelt-Fe-atom…Identifikationen af den elektroniske og strukturelle dynamik af katalytiske centre i enkelt-Fe-atom materiale ved Operando Mossbauer spektroskopi. Kredit:LI Xuning Single-atom catalyst (SAC) er
Forskere identificerer elektroniske og strukturelle dynamikker i katalytiske centre i enkelt-Fe-atom…Identifikationen af den elektroniske og strukturelle dynamik af katalytiske centre i enkelt-Fe-atom materiale ved Operando Mossbauer spektroskopi. Kredit:LI Xuning Single-atom catalyst (SAC) er -
 Bifile overflader reducerer afrimningstider i varmevekslereTime-lapse billeder af dynamisk afrimning på superhydrofobe og bifile overflader. Tidspunktet t =0 repræsenterer det tidspunkt, hvor smeltning af frost først blev observeret visuelt. Kredit:Nenad Milj
Bifile overflader reducerer afrimningstider i varmevekslereTime-lapse billeder af dynamisk afrimning på superhydrofobe og bifile overflader. Tidspunktet t =0 repræsenterer det tidspunkt, hvor smeltning af frost først blev observeret visuelt. Kredit:Nenad Milj -
 Udskrivning flader polymerer, forbedring af elektriske og optiske egenskaberProfessor Ying Diao, venstre, postdoc forsker Kyung Sun Park, siddende, og kandidatstuderende Justin Kwok har fundet ud af, at snoede polymerer kan flades ud via printprocessen for at gøre dem bedre t
Udskrivning flader polymerer, forbedring af elektriske og optiske egenskaberProfessor Ying Diao, venstre, postdoc forsker Kyung Sun Park, siddende, og kandidatstuderende Justin Kwok har fundet ud af, at snoede polymerer kan flades ud via printprocessen for at gøre dem bedre t -
 Proteinmiljøet gør katalysatoren effektiv til brintproduktionBochum -forskerne Martin Winkler, Oliver Lampret og Thomas Happe (fra venstre mod højre) sammen med Olaf Rüdiger (i baggrunden) fra Max Planck Institute. Kredit:RUB, Marquard Interaktionen mellem
Proteinmiljøet gør katalysatoren effektiv til brintproduktionBochum -forskerne Martin Winkler, Oliver Lampret og Thomas Happe (fra venstre mod højre) sammen med Olaf Rüdiger (i baggrunden) fra Max Planck Institute. Kredit:RUB, Marquard Interaktionen mellem
- Mere store, højintensive skovbrande sandsynligvis i de kommende år
- Sådan beregnes effektiviteten af en elektrisk generator
- Undersøgelse finder disciplinforskelle i børnehave drevet af racemæssig skævhed
- Ryanair advarede om at respektere den nationale arbejdslovgivning i Europa
- Intel introducerer kryogen kontrolchip Horse Ridge for at muliggøre styring af flere kvantebits
- Undersøgelse af gamle klipper tyder på iltsvind i havene førte til ende-trias masseudryddelse