


Simuleringer giver kort til skattekammeret af fluorerede forbindelser
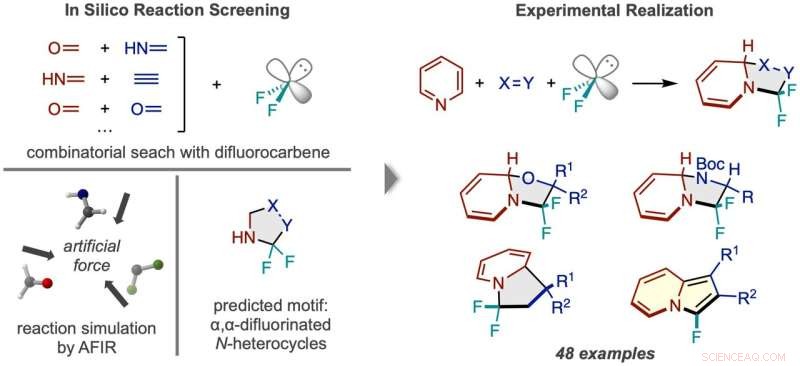
Workflow af reaktionsopdagelse via in silico screening. (Venstre) Reaktioner mellem difluorcarben og adskillige par af små molekyler blev simuleret, hvilket forudsagde et N-heterocyklisk produkt fluoreret to gange ved alfa-carbonet. (Til højre) Den vellykkede reaktionsramme ved hjælp af pyridin og eksempler på de opnåede typer af produktforbindelser. Kredit:Nature Synthesis (2022). DOI:10.1038/s44160-022-00128-y
Computersimuleringer bruges oftest som en guide, så kemikere mere effektivt kan finde ud af de nøjagtige detaljer i en generel reaktionsidé, de har i tankerne - ligesom et kompas hjælper med at guide en opdagelsesrejsende effektivt til en destination på deres kort. Forskere ved ICReDD tog imidlertid tingene et stort skridt videre og brugte simuleringer til at producere den generelle idé til en helt uanet reaktion, ved effektivt at bruge beregninger til at lave selve kortet. Ved at bruge designprincippet foreslået af beregningsresultater, ramte holdet moderlodden i laboratoriet og udviklede med succes en række af 48 reaktioner, der producerer forbindelser, der potentielt er nyttige til udvikling af nye lægemidler.
Tilstedeværelsen og positionen af fluor i et molekyle påvirker ofte et molekyles farmakologiske aktivitet. Forskere ved ICReDD har brugt kvantekemiske beregninger til at opdage en reaktion, der selektivt tilføjer to fluoratomer til en vanskelig tilgængelig position på en N-heterocyklus-molekyler med en kulstofringstruktur, hvor mindst ét kulstof i ringen er erstattet med nitrogen . Evnen til at binde fluoratomer til det tidligere svært tilgængelige "alfa-carbon" - kulstoffet umiddelbart ved siden af nitrogenet i ringstrukturen - kan føre til udviklingen af en lang række nye lægemidler.
Inden forskerne udførte eksperimenter i laboratoriet, kastede forskerne et bredt net og testede beregningsmæssigt levedygtigheden af adskillige 3-komponent reaktioner ved hjælp af den kunstige kraftinducerede reaktion (AFIR) metode. De simulerede reaktionen af et difluorcarben-molekyle, som virker ved kilden til fluoratomer, med forskellige par af små molekyler med en dobbelt- eller tredobbeltbinding. Disse simuleringer viste, at en række ringdannende reaktioner burde være levedygtige.
Forskere prøvede en af de lovende reaktioner, der blev foreslået af de indledende beregningsresultater, men lykkedes ikke. A more narrowly focused, optimized computation of the transition state energy of the reaction in question showed that the difluorocarbene molecule more easily reacted with itself than with the desired starting molecules, signaling that an undesired side reaction was likely occurring. This result inspired researchers to change one of the starting materials to the cyclic molecule pyridine, which they anticipated would be able to compete with the unwanted side reaction. This change resulted in the successful synthesis of the desired N-heterocyclic product with two fluorines attached at the alpha carbon position.
The reaction developed here is also significant because it breaks the aromatic system of electrons in the pyridine molecule, a transformation that is especially difficult due to the high stability of aromatic systems. Additionally, the 3-component reaction framework was applied successfully in the lab to a wide range of starting materials, resulting in many new molecules with unique alpha position fluorine substitutions. The large scope of reactivity greatly increases the potential utility of this reaction framework in new drug development.
The researchers see their streamlined screening method as a way to broaden the scope of their search and discover new horizons in chemical reaction design.
"Our study's highlight is the successful demonstration of an in silico reaction screening strategy for reaction development. The computational reaction simulation suggested less-explored three-component reactions of difluorocarbene and two unsaturated molecules, which we successfully realized in experiments," explained lead author Hiroki Hayashi.
"I think the AFIR method is a powerful tool for dictating new research directions in reaction discovery, and we plan to continue building a computation-based reaction development platform by integrating the computational and informatics techniques of ICReDD."
The study was published in Nature Synthesis . + Udforsk yderligere
Hitting rewind to predict multi-step chemical reactions
 Varme artikler
Varme artikler
-
 Nye gelbelægninger kan føre til bedre katetre og kondomerMIT ingeniører har designet et gel-lignende materiale, der kan belægges på standard plast- eller gummienheder, giver en blødere, mere glat ydre, der betydeligt kan lette en patients ubehag under opera
Nye gelbelægninger kan føre til bedre katetre og kondomerMIT ingeniører har designet et gel-lignende materiale, der kan belægges på standard plast- eller gummienheder, giver en blødere, mere glat ydre, der betydeligt kan lette en patients ubehag under opera -
 Efterligner enzymer, kemikere producerer store, nyttige carbonringeMakrocykliseringsstrategier. (A) Tidligere tilgange og foldamertilgang til makrocyklisering. (B) Divergent reaktivitet:Foldamer versus små molekyle katalyse. lign., ækvivalent(er). Kredit: Videnskab
Efterligner enzymer, kemikere producerer store, nyttige carbonringeMakrocykliseringsstrategier. (A) Tidligere tilgange og foldamertilgang til makrocyklisering. (B) Divergent reaktivitet:Foldamer versus små molekyle katalyse. lign., ækvivalent(er). Kredit: Videnskab -
 Forskere udvikler biogummilim til hurtigere kirurgisk bedring og smertelindringFlydende CaproGlu påført kød, der er hærdet med UV-lys, bliver til biogummi. Kredit:Nanyang Technological University Materialeforskere fra Nanyang Technological University, Singapore (NTU Singapor
Forskere udvikler biogummilim til hurtigere kirurgisk bedring og smertelindringFlydende CaproGlu påført kød, der er hærdet med UV-lys, bliver til biogummi. Kredit:Nanyang Technological University Materialeforskere fra Nanyang Technological University, Singapore (NTU Singapor -
 Lovende anticancer-molekyle identificeretFigur 1. En integreret tilgang til HS-AFM og molekylær docking for at afsløre bindingsmekanismen for Apt-7 til CYP24 (A) Den tredimensionelle visning af de toprangerede dockede konformationer af CYP24
Lovende anticancer-molekyle identificeretFigur 1. En integreret tilgang til HS-AFM og molekylær docking for at afsløre bindingsmekanismen for Apt-7 til CYP24 (A) Den tredimensionelle visning af de toprangerede dockede konformationer af CYP24
- En super-alge til at redde vores have
- Det største antikke DNA-studie nogensinde belyser årtusinder af syd- og centralasiatisk forhistori…
- Et skridt foran i kapløbet mod ultrahurtig billeddannelse af enkeltpartikler
- Konstrueret multivalent selvsamlet bindeprotein mod SARS-CoV-2 RBD
- To planeter i kredsløb om nærliggende stjerne opdaget med TESS
- Hvordan cryptocurrency -diskussioner spredes