


Machine learning framework ID'er mål til forbedring af katalysatorer
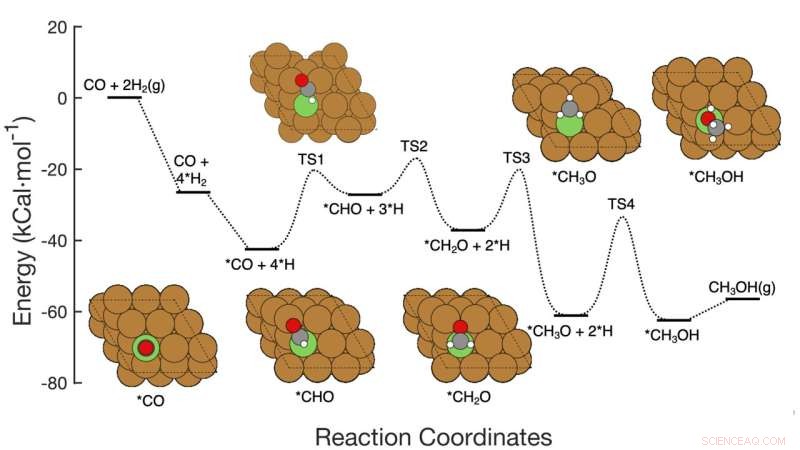
Denne grafik viser syvtrins reaktionsvejen for CO-hydrogenering til methanol over kobberbaserede katalysatorer, inklusive reaktanterne på hvert trin, skematiske atomarrangementer af mellemprodukterne og de energiaktiveringsbarrierer, der kræves for at komme fra trin til trin. Brookhaven Lab-teamet demonstrerede en maskinlæringsramme, der med succes identificerede, hvilke trin/kombinationer af trin, der skulle justeres for at forbedre methanolproduktionen. Deres arbejde kunne hjælpe med at guide designet af nye katalysatorer for at nå dette mål, og rammerne kan anvendes til at optimere andre reaktioner. Kredit:Brookhaven National Laboratory
Kemikere ved det amerikanske energiministeriums Brookhaven National Laboratory har udviklet en ny maskinlæringsramme (ML), der kan nulstille hvilke trin i en flertrins kemisk konvertering, der skal justeres for at forbedre produktiviteten. Tilgangen kunne hjælpe med at guide designet af katalysatorer - kemiske "dealmakers", der fremskynder reaktioner.
Holdet udviklede metoden til at analysere omdannelsen af carbonmonoxid (CO) til methanol ved hjælp af en kobberbaseret katalysator. Reaktionen består af syv ret ligetil elementære trin.
"Vores mål var at identificere, hvilket elementært trin i reaktionsnetværket eller hvilken delmængde af trin, der styrer den katalytiske aktivitet," sagde Wenjie Liao, den første forfatter på et papir, der beskriver metoden, der netop er offentliggjort i tidsskriftet Catalysis Science &Technology . Liao er en kandidatstuderende ved Stony Brook University, som har arbejdet med forskere i Catalysis Reactivity and Structure (CRS)-gruppen i Brookhaven Labs kemidivision.
Ping Liu, CRS-kemikeren, der ledede arbejdet, sagde:"Vi brugte denne reaktion som et eksempel på vores ML-rammemetode, men du kan sætte enhver reaktion ind i denne ramme generelt."
Målretning mod aktiveringsenergier
Forestil dig en kemisk reaktion i flere trin som en rutsjebane med bakker i forskellige højder. Højden af hver bakke repræsenterer den energi, der er nødvendig for at komme fra det ene trin til det næste. Katalysatorer sænker disse "aktiveringsbarrierer" ved at gøre det lettere for reaktanter at komme sammen eller ved at lade dem gøre det ved lavere temperaturer eller tryk. For at fremskynde den overordnede reaktion skal en katalysator målrette det eller de trin, der har den største effekt.
Traditionelt ville forskere, der søger at forbedre en sådan reaktion, beregne, hvordan man ændrer hver aktiveringsbarriere en ad gangen kan påvirke den samlede produktionshastighed. Denne type analyse kunne identificere, hvilket trin der var "hastighedsbegrænsende", og hvilke trin der bestemmer reaktionsselektiviteten - det vil sige om reaktanterne fortsætter til det ønskede produkt eller ad en alternativ vej til et uønsket biprodukt.

Brookhaven Lab kemiker Ping Liu og Wenjie Liao, en kandidatstuderende ved Stony Brook University, udviklede en maskinlæringsramme for at identificere, hvilke kemiske reaktionstrin der kunne målrettes for at forbedre reaktionsproduktiviteten. Kredit:Brookhaven National Laboratory
Men ifølge Liu, "disse estimater ender med at være meget grove med en masse fejl for nogle grupper af katalysatorer. Det har virkelig gjort ondt for katalysatordesign og -screening, hvilket er det, vi forsøger at gøre," sagde hun.
Den nye maskinlæringsramme er designet til at forbedre disse estimater, så forskerne bedre kan forudsige, hvordan katalysatorer vil påvirke reaktionsmekanismer og kemisk output.
"Nu, i stedet for at flytte én barriere ad gangen, flytter vi alle barriererne samtidigt. Og vi bruger maskinlæring til at fortolke det datasæt," sagde Liao.
Denne tilgang, sagde teamet, giver meget mere pålidelige resultater, herunder om hvordan trin i en reaktion arbejder sammen.
"Under reaktionsbetingelser er disse trin ikke isoleret eller adskilt fra hinanden; de er alle forbundet," sagde Liu. "Hvis du bare gør et trin ad gangen, går du glip af en masse information - interaktionerne mellem de elementære trin. Det er det, der er blevet fanget i denne udvikling," sagde hun.
Opbygning af modellen
Forskerne startede med at bygge et datasæt for at træne deres maskinlæringsmodel. Datasættet var baseret på "density functional theory" (DFT) beregninger af den aktiveringsenergi, der kræves for at transformere et arrangement af atomer til det næste gennem reaktionens syv trin. Derefter kørte forskerne computerbaserede simuleringer for at udforske, hvad der ville ske, hvis de ændrede alle syv aktiveringsbarrierer samtidigt – nogle går op, nogle går ned, nogle individuelt og nogle i par.
"Rækken af data, vi inkluderede, var baseret på tidligere erfaringer med disse reaktioner og dette katalytiske system inden for det interessante variationsområde, der sandsynligvis vil give dig bedre ydeevne," sagde Liu.
Ved at simulere variationer i 28 "deskriptorer" - inklusive aktiveringsenergierne for de syv trin plus par af trin, der skiftede to ad gangen - producerede teamet et omfattende datasæt med 500 datapunkter. Dette datasæt forudsagde, hvordan alle disse individuelle tweaks og par af tweaks ville påvirke methanolproduktionen. Modellen scorede derefter de 28 deskriptorer efter deres betydning for at drive methanoloutput.
"Vores model 'lærte' af dataene og identificerede seks nøgledeskriptorer, som den forudsiger ville have størst indflydelse på produktionen," sagde Liao.
Efter at de vigtige deskriptorer var blevet identificeret, omskolede forskerne ML-modellen ved at bruge blot de seks "aktive" deskriptorer. Denne forbedrede ML-model var i stand til at forudsige katalytisk aktivitet udelukkende baseret på DFT-beregninger for disse seks parametre.
"I stedet for at du skal beregne hele 28 deskriptorer, kan du nu kun beregne med de seks deskriptorer og få de methanolkonverteringsrater, du er interesseret i," sagde Liu.
Holdet siger, at de også kan bruge modellen til at screene katalysatorer. Hvis de kan designe en katalysator, der forbedrer værdien af de seks aktive deskriptorer, forudsiger modellen en maksimal methanolproduktionshastighed.
Forståelse af mekanismer
Da holdet sammenlignede forudsigelserne af deres model med den eksperimentelle ydeevne af deres katalysator - og ydeevnen af legeringer af forskellige metaller med kobber - stemte forudsigelserne overens med de eksperimentelle resultater. Sammenligninger af ML-tilgangen med den tidligere metode, der blev brugt til at forudsige legerings ydeevne, viste, at ML-metoden var langt overlegen.
Dataene afslørede også en masse detaljer om, hvordan ændringer i energibarrierer kunne påvirke reaktionsmekanismen. Af særlig interesse - og vigtig - var, hvordan forskellige trin i reaktionen arbejder sammen. For eksempel viste dataene, at i nogle tilfælde ville en sænkning af energibarrieren i det hastighedsbegrænsende trin alene ikke i sig selv forbedre methanolproduktionen. Men at justere energibarrieren for et trin tidligere i reaktionsnetværket, samtidig med at aktiveringsenergien for det hastighedsbegrænsende trin holdes inden for et ideelt område, ville øge methanoloutputtet.
"Vores metode giver os detaljerede oplysninger, som vi måske kan bruge til at designe en katalysator, der koordinerer interaktionen mellem disse to trin godt," sagde Liu.
Men Liu er mest begejstret for potentialet for at anvende sådanne datadrevne ML-rammer til mere komplicerede reaktioner.
"Vi brugte methanol-reaktionen til at demonstrere vores metode. Men den måde, den genererer databasen på, og hvordan vi træner ML-modellen, og hvordan vi interpolerer rollen af hver deskriptors funktion for at bestemme den samlede vægt i forhold til deres betydning - det kan være anvendes let på andre reaktioner," sagde hun. + Udforsk yderligere
Opdagelse af en ny katalysator til højaktiv og selektiv carbondioxidhydrogenering til methanol
Sidste artikelNy forståelse af nøglebrændselscellekatalysator
Næste artikelSkjulte forvrængninger udløser lovende termoelektrisk egenskab
 Varme artikler
Varme artikler
-
 Begrænsninger af modeller i videnskabEn model er en beskrivelse af naturfænomenet, som forskere kan bruge til at fremsætte forudsigelser. En god model er både så nøjagtig som muligt og så enkel som muligt, hvilket gør den ikke kun kra
Begrænsninger af modeller i videnskabEn model er en beskrivelse af naturfænomenet, som forskere kan bruge til at fremsætte forudsigelser. En god model er både så nøjagtig som muligt og så enkel som muligt, hvilket gør den ikke kun kra -
 Reduktion af CO2 med fælles elementer og sollysCO 2 reduktion ved anvendelse af en fotokatalysator, der kombinerer carbonnitrid og et jernkompl. Kredit:Osamu Ishitani En international forskningsgruppe, der samarbejder med Tokyo Institute of
Reduktion af CO2 med fælles elementer og sollysCO 2 reduktion ved anvendelse af en fotokatalysator, der kombinerer carbonnitrid og et jernkompl. Kredit:Osamu Ishitani En international forskningsgruppe, der samarbejder med Tokyo Institute of -
 Gamle enzymer katalysatorer for nye opdagelser(L-R) Elizabeth Gillam og Jong-Min (Joseph) Baek ser på P450 enzymspektre på et specialiseret spektrofotometer, vurdere om proteinet er intakt eller inaktivt. Kredit:University of Queensland Forsk
Gamle enzymer katalysatorer for nye opdagelser(L-R) Elizabeth Gillam og Jong-Min (Joseph) Baek ser på P450 enzymspektre på et specialiseret spektrofotometer, vurdere om proteinet er intakt eller inaktivt. Kredit:University of Queensland Forsk -
 Hvordan to vandmolekyler danser sammenKredit:CC0 Public Domain Et internationalt forskerhold har fået ny indsigt i, hvordan vandmolekyler interagerer. En laser med særlig høj lysstyrke, som findes på FELIX-laboratoriet på Radboud Univ
Hvordan to vandmolekyler danser sammenKredit:CC0 Public Domain Et internationalt forskerhold har fået ny indsigt i, hvordan vandmolekyler interagerer. En laser med særlig høj lysstyrke, som findes på FELIX-laboratoriet på Radboud Univ
- Eksperter i Antarktis tilbyder to mulige udsigter over fremtidens kontinenter
- Model hjælper robotter med at navigere mere som mennesker gør
- Fleksibel, stabil og potent mod kræft - ny tilgang til tumorbehandling
- NASA ser på koncentrationen af orkanen Helenes vanddamp
- Belo Monte:Der er intet grønt eller bæredygtigt ved disse mega-dæmninger
- Landdistrikter og bysamfund har brug for forskellige politikker for at øge den økonomiske mobilite…